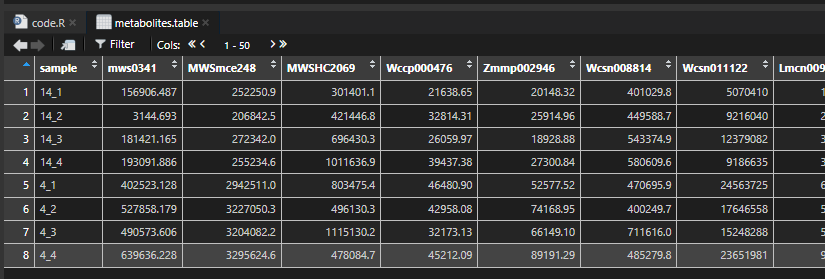
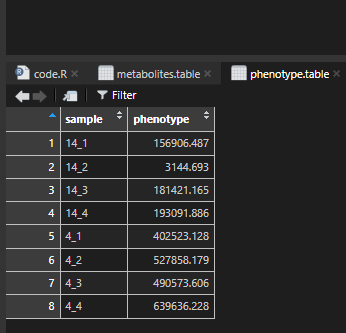
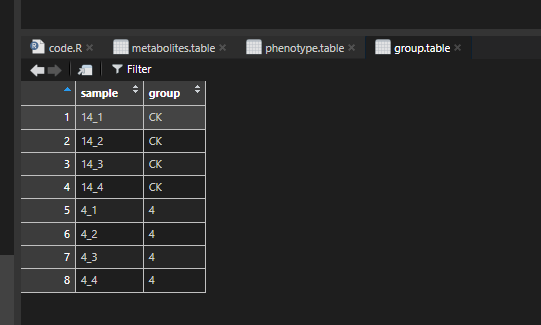
callDAMs(metabolites.table = metabolites.table,
phenotype.table = phenotype.table,
group.table = group.table,
use.lm = TRUE,
use.cor = TRUE,
lm.R2 = 0.5) -> lx
#' @name callDAMs
#' @author Xiang LI <lixiang117423@@foxmail.com>
#'
#' @title DAMs using multiple methods.
#'
#' @export
#'
# load my functions
# ggplot2 theme
callDAMs <- function(metabolites.table,
phenotype.table,
group.table,
CK.group = "CK",
use.oplsda = TRUE,
use.log2FC = TRUE,
use.wilcox = TRUE,
use.DESeq2 = TRUE,
use.lm = FALSE,
use.cor = FALSE,
VIP.value = 1,
log2FC.value.abs = 1,
Wilcokx.pvalue = 0.05,
DESeq2.log2FC.abs = 1,
DESeq2.pvalue = 0.05,
DESeq2.qvalue = 0.05,
lm.R2 = 0.9,
lm.pvalue = 0.05,
cor.method = "pearson",
cor.value.abs = 0.6,
cor.pvalue = 0.05) {
# OPLS-DA
group.table$group %>%
factor(levels = c("CK", setdiff(unique(group.table$group), "CK"))) -> group
metabolites.table %>%
tibble::column_to_rownames(var = "sample") -> df.meta
ropls::opls(df.meta, group, crossvalI = 6, predI = 2, orthoI = 1) -> opls.da
opls.da@vipVn %>%
as.data.frame() %>%
magrittr::set_names(c("VIP")) %>%
dplyr::filter(VIP >= VIP.value) %>%
tibble::rownames_to_column(var = "Index") %>%
dplyr::rename(value = VIP) %>%
dplyr::mutate(method = "OPLS-DA") -> df.vip
# log2FC
metabolites.table %>%
tidyr::pivot_longer(cols = 2:ncol(.)) %>%
dplyr::left_join(group.table) %>%
dplyr::group_by(group, name) %>%
dplyr::summarise(mean.value = mean(value)) %>%
dplyr::ungroup() %>%
tidyr::pivot_wider(names_from = group, values_from = mean.value) %>%
dplyr::select(name, CK, setdiff(unique(group.table$group), "CK")) %>%
magrittr::set_names(c("Index", "CK", "treatment")) %>%
dplyr::mutate(log2FC = (treatment / CK) %>% log2()) %>%
dplyr::select(Index, log2FC) %>%
dplyr::filter(abs(log2FC) > log2FC.value.abs) -> df.log2FC
# Wilcox
metabolites.table %>%
tidyr::pivot_longer(cols = 2:ncol(.)) %>%
dplyr::left_join(group.table) %>%
dplyr::group_by(name) %>%
rstatix::wilcox_test(value ~ group) %>%
dplyr::ungroup() %>%
dplyr::filter(p < Wilcokx.pvalue) %>%
dplyr::select(name, p) %>%
magrittr::set_names(c("Index", "Wilcox.pvalue")) -> df.wilcox
# DESeq2
metabolites.table %>%
tibble::column_to_rownames(var = "sample") %>%
t() %>%
as.data.frame() %>%
round(0) -> df.meta
group.table %>%
dplyr::mutate(group = factor(group, level = c("CK", setdiff(unique(group.table$group), "CK")))) %>%
tibble::column_to_rownames(var = "sample") -> df.group
DESeq2::DESeqDataSetFromMatrix(
countData = df.meta,
colData = df.group,
design = ~ group
) %>%
DESeq2::DESeq() %>%
DESeq2::results() %>%
as.data.frame() %>%
dplyr::mutate(signif.group = case_when(
log2FoldChange > DESeq2.log2FC.abs & pvalue < DESeq2.pvalue ~ "Up",
log2FoldChange < -DESeq2.log2FC.abs & pvalue < DESeq2.pvalue ~ "Down",
TRUE ~ "NS"
)) %>%
tibble::rownames_to_column(var = "Index") %>%
dplyr::filter(abs(log2FoldChange) > DESeq2.log2FC.abs, pvalue < DESeq2.pvalue, padj < DESeq2.qvalue) -> df.deseq2
if (use.cor) {
# 相关性
metabolites.table %>%
tibble::column_to_rownames(var = "sample") -> df.meta
phenotype.table %>%
tibble::column_to_rownames(var = "sample") -> df.phenotype
WGCNA::corAndPvalue(df.meta, df.phenotype, method = cor.method) -> cor.p
cor.p$cor %>%
as.data.frame() %>%
magrittr::set_names("cor") %>%
tibble::rownames_to_column(var = "Index") -> df.cor
cor.p$p %>%
as.data.frame() %>%
magrittr::set_names("p") %>%
tibble::rownames_to_column(var = "Index") %>%
dplyr::left_join(df.cor) %>%
dplyr::select(Index, cor, p) %>%
dplyr::filter(abs(cor) > cor.value.abs, p < cor.pvalue)-> df.cor.p
# Intersection
intersect(df.vip$Index, df.log2FC$Index) %>%
intersect(df.wilcox$Index) %>%
intersect(df.deseq2$Index) %>%
intersect(df.cor.p$Index) -> df.intersection.1
}else{
df.cor.p = NULL
# Intersection
intersect(df.vip$Index, df.log2FC$Index) %>%
intersect(df.wilcox$Index) %>%
intersect(df.deseq2$Index) -> df.intersection.1
}
if (use.lm) {
# lm
metabolites.table %>%
tidyr::pivot_longer(cols = 2:ncol(.)) %>%
dplyr::left_join(phenotype.table) -> df.tmp
all.lm = NULL
for (i in unique(df.tmp$name)) {
df.tmp %>%
dplyr::filter(name == i) -> df.tmp.2
lm(value ~ phenotype, data = df.tmp.2) -> fit
}
data.frame(R2 = summary(fit)$adj.r.squared %>% round(3),
p.model = anova(fit)$`Pr(>F)`[1]) %>%
dplyr::mutate(Index = i) %>%
dplyr::select(Index, R2, p.model) %>%
dplyr::bind_rows(all.lm) -> all.lm
all.lm %>%
dplyr::filter(R2 > lm.R2, p.model < lm.pvalue) -> df.lm
intersect(df.intersection.1, df.lm$Index) -> df.intersection.2
}else{
df.lm = NULL
df.intersection.1 -> df.intersection.2
}
# # Intersection
# intersect(df.vip$Index, df.log2FC$Index) %>%
# intersect(df.wilcox$Index) %>%
# intersect(df.deseq2$Index) %>%
# intersect(df.cor.p$Index) %>%
# intersect(df.lm$Index) -> df.intersection
#
# 返回结果
reture.list = list(OPLS.DA = df.vip,
log2FC = df.log2FC,
Wilcox = df.wilcox,
DESeq2 = df.deseq2,
cor = df.cor.p,
lm = df.lm,
intersection = df.intersection.2
)
return(reture.list)
}



