写在前面
趁着这次培训,把学的东西都整理一下。不断更新,想到啥更新啥。。。。。。
软件推荐
ssh工具:目前在用的是Tabby,比较满意的是在一个窗口就可以实现ssh+文件的上传下载。markdown工具:现在在用的是Typora,付费版本。代码编辑工具:
-R:RStudio- 其他:
VS Code
- 其他:
软件安装
VScode
使用VScode主要是为了方便远程连接服务器进行远程开发。体验了几次,还是很爽的。
- 安装Remote-SSH
- 设置配置文件:输入服务器名称、IP地址、端口和用户名即可。
- 更改设置:更改设置,
File->Preferences->Settings->Extension->Remote-SSH,找到 Show Login Terminal 并勾选。
后续提示输入密码,然后就可以 登录使用了。代码编辑器和终端在一个页面,就很方便。
conda
初学生信最折腾的就是软件安装,现在我基本上都是用mamba进行安装。mamba本质是conda,但是解析速度非常快,下载速度也非常快。

现在直接从官方网站下载即可安装,之前还需要先安装conda,官方也不推荐使用conda安装mamba.
以Ubuntu系统为例:
wget "https://github.com/conda-forge/miniforge/releases/latest/download/Mambaforge-$(uname)-$(uname -m).sh"
bash Mambaforge-$(uname)-$(uname -m).sh- 创建环境(假如需要指定
python版本):
mamba create --name 环境名称 python=3.11- 激活环境
mamba activate 环境名称- 添加
channels
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/main/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge/
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda/- 查看环境都有哪些
mamba env list- 安装软件:我通常是直接去ANACONDA上搜,比如
fastqc:
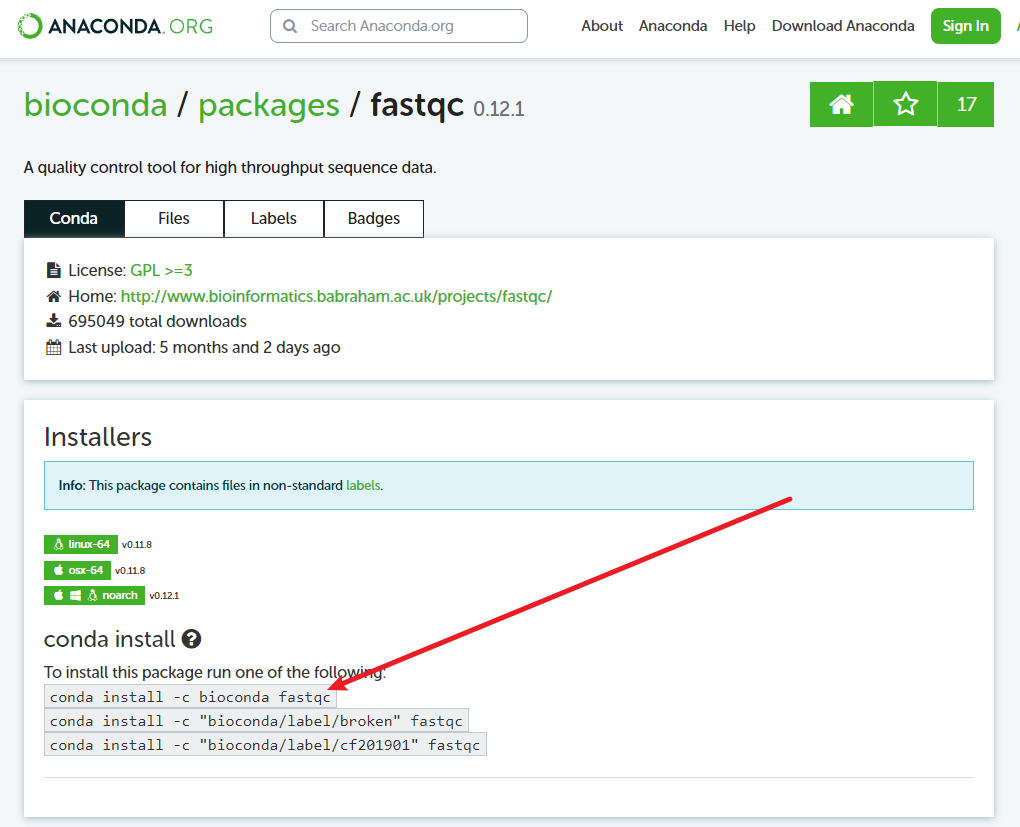
把conda改成mamba就行:
mamba install -c bioconda fastqc- 不更新依赖安装:某些大项目往往需要很多软件,各个软件依赖的其他软件版本通常不一样,这个时候可以考虑不更新依赖进行安装:
mamba install -c bioconda fastqc --freeze-installed其他不能用mamba安装的软件就只能根据官方文档进行安装了。
Docker
在Ubuntu上安装Docker
# Add Docker's official GPG key:
sudo apt-get update
sudo apt-get install ca-certificates curl gnupg
sudo install -m 0755 -d /etc/apt/keyrings
curl -fsSL https://download.docker.com/linux/ubuntu/gpg | sudo gpg --dearmor -o /etc/apt/keyrings/docker.gpg
sudo chmod a+r /etc/apt/keyrings/docker.gpg
# Add the repository to Apt sources:
echo \
"deb [arch="$(dpkg --print-architecture)" signed-by=/etc/apt/keyrings/docker.gpg] https://download.docker.com/linux/ubuntu \
"$(. /etc/os-release && echo "$VERSION_CODENAME")" stable" | \
sudo tee /etc/apt/sources.list.d/docker.list > /dev/null
sudo apt-get update
sudo apt-get install docker-ce docker-ce-cli containerd.io docker-buildx-plugin docker-compose-plugin将用户加入docker用户组:
sudo usermod -aG docker username查看哪些用户在docker用户组中:
grep '^docker:' /etc/group将特定的目录挂载的到docker的工作目录下:
docker run -it --name cactus.test -v ~/lixiang/yyt.acuce.3rd:/data quay.io/comparative-genomics-toolkit/cactus:v2.6.11docker的默认目录是/data,-v参数将~/lixiang/yyt.acuce.3rd目录挂载到/data目录下。
输入exit退出当前容器。使用docker rm name删除对应的容器名称。
文件格式
gVCF
表头都有详细的注释信息,正文部分如下:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1
Chr1 1 . A <*> 0 . END=914 GT:GQ:MIN_DP:PL 0/0:1:0:0,0,0
Chr1 915 . T <*> 0 . END=915 GT:GQ:MIN_DP:PL ./.:0:1:29,3,0
Chr1 916 . A <*> 0 . END=977 GT:GQ:MIN_DP:PL 0/0:1:0:0,3,29
Chr1 978 . A <*> 0 . END=978 GT:GQ:MIN_DP:PL ./.:0:1:29,3,0每一列的含义如下:
- #CHROM: 染色体的名字。
- POS: 变异发生的位置。
- ID: 变异的唯一标识符,通常为 “.” 表示未知或不适用。
- REF: 参考基因的碱基。
- ALT: 变异的碱基,可能是一个单一的碱基、多个碱基(例如插入或删除)、或者用
<*>表示未知。 - QUAL: 变异的质量得分。
- FILTER: 变异是否通过过滤器,通常为 “.” 表示未经过滤。
- INFO: 包含更多关于变异的信息,如 END=977 表示变异的结束位置。
- FORMAT: 描述了样本基因型信息的格式。
- Sample1: 实际样本的基因型信息,如 “0/0:1:0:0,0,0” 表示该样本为纯合子(两个相同的等位基因),具体基因型信息依赖于 FORMAT 中的描述。
在示例中,可以看到有两个样本(Sample1 和 ./.),每个样本有对应的基因型信息。纯合子通常表示为 “0/0”,而缺失数据通常表示为 “./.”。
要查看 VCF 文件,可以使用文本编辑器或者专用的 VCF 查看工具。如果使用文本编辑器,每一行的不同字段通过制表符或空格分隔。
在线数据库
- 33个水稻高质量基因组数据库(四川农大):Rice Resource Center
本地数据库
套用的数据库在本节介绍安装配置方法,其他专业的数据库会对应章节介绍。
NCBI
截至2023年8月8日,最新版本是V5,需要修改下方代码。
nt
下载NSBI官方构建好索引的数据库,使用ascp加速下载,下载完成直接解压就能用。
mkcd ~/database/ncbi.nt
~/.aspera/connect/bin/ascp -i ~/mambaforge/envs/tools4bioinf/etc/asperaweb_id_dsa.openssh --overwrite=diff -QTr -l6000m \
anonftp@ftp.ncbi.nlm.nih.gov:blast/db/v4/nt_v4.{00..85}.tar.gz ./nr
下载NSBI官方构建好索引的数据库,使用ascp加速下载,下载完成直接解压就能用。
mkcd ~/database/ncbi.nr
~/.aspera/connect/bin/ascp -i ~/mambaforge/envs/tools4bioinf/etc/asperaweb_id_dsa.openssh --overwrite=diff -QTr -l6000m \
anonftp@ftp.ncbi.nlm.nih.gov:blast/db/v4/nt_v4.{00..79}.tar.gz ./taxonomy
mkcd ~/database/ncbi.taxonomy
axel -n 50 ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz
tar -xvf taxdump.tar.gzCDD
NCBI-CDD用于结构域检索和验证,官方网站一次只能上传4000条序列,有时候比较影响进度,下面的代码用于本地数据库构建和比对。
# 下载数据库数据
ascp -v -k 1 -T -l 1000m -i ~/mambaforge/envs/tools4bioinf/etc/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nih.gov:/pub/mmdb/cdd/cdd.tar.gz ./
# 构建数据库
makeprofiledb -in Cdd.pn -out ../db/ncbi.cdd -dbtype rps
# 比对
rpsblast -query results/oryza.1/6.all.wrky.pep.fa -outfmt 6 -evalue 0.01 -db ~/database/ncbi.cdd/db/ncbi.cdd -out results/oryza.1/7.ncbi.cdd.res.txt -num_threads 60
# 下载分类信息
ascp -v -k 1 -T -l 1000m -i ~/mambaforge/envs/tools4bioinf/etc/asperaweb_id_dsa.openssh anonftp@ftp.ncbi.nih.gov:/pub/mmdb/cdd/cddid.tbl.gz ./ 示意图绘制
生物序列展示
Illustrator for Biological Sequences
Linux基础
命令行界面配置
有个好看的界面,在报错的时候心情会好一些:
zsh配置文件.zshrc配置细节(uername改成自己的用户名):
###################################################################################################
########################################## 默认设置 ################################################
###################################################################################################
# Enable Powerlevel10k instant prompt. Should stay close to the top of ~/.zshrc.
# Initialization code that may require console input (password prompts, [y/n]
# confirmations, etc.) must go above this block; everything else may go below.
if [[ -r "${XDG_CACHE_HOME:-$HOME/.cache}/p10k-instant-prompt-${(%):-%n}.zsh" ]]; then
source "${XDG_CACHE_HOME:-$HOME/.cache}/p10k-instant-prompt-${(%):-%n}.zsh"
fi
# Path to your oh-my-zsh installation.
export ZSH="/home/username/.oh-my-zsh"
# tabby ftp工具默认当前位置
precmd () { echo -n "\x1b]1337;CurrentDir=$(pwd)\x07" }
#ZSH_THEME="robbyrussell"
#ZSH_THEME="powerlevel10k/powerlevel10k"
# Add wisely, as too many plugins slow down shell startup.
plugins=(git)
source $ZSH/oh-my-zsh.sh
# To customize prompt, run `p10k configure` or edit ~/.p10k.zsh.
[[ ! -f ~/.p10k.zsh ]] || source ~/.p10k.zsh
###################################################################################################
########################################## conda/mamba初始化 ######################################
###################################################################################################
# >>> conda initialize >>>
# !! Contents within this block are managed by 'conda init' !!
__conda_setup="$('/home/username/mambaforge/bin/conda' 'shell.zsh' 'hook' 2> /dev/null)"
if [ $? -eq 0 ]; then
eval "$__conda_setup"
else
if [ -f "/home/username/mambaforge/etc/profile.d/conda.sh" ]; then
. "/home/username/mambaforge/etc/profile.d/conda.sh"
else
export PATH="/home/username/mambaforge/bin:$PATH"
fi
fi
unset __conda_setup
if [ -f "/home/username/mambaforge/etc/profile.d/mamba.sh" ]; then
. "/home/username/mambaforge/etc/profile.d/mamba.sh"
fi
# <<< conda initialize <<<
LD_LIBRARY_PATH=$LD_LIBRARY_PATH:/home/username/mambaforge/lib
export LD_LIBRARY_PATH
export PATH=$PATH:/home/username/software/mira/bin
###################################################################################################
########################################### 快捷键 ################################################
###################################################################################################
alias np='nohup'
alias tt='tail'
alias np='nohup'
alias pp='_pp(){ps -aux | grep $1};_pp'
alias rf='rm -rf'
alias mm='mamba'
alias mma='mamba activate'
alias mmd='mamba deactivate'
alias mml='mamba env list'
alias hg='_hg(){history | grep $1 | tail -n $2};_hg'
alias hh='htop'
alias mmt='mamba activate tools4bioinf'
alias pp='python'
alias hh='head'
alias get='axel -n 30'
alias gh='_gh(){grep ">" $1 | head};_gh'
alias pp='scp -r ubuntu@43.153.77.165:/home/ubuntu/download/\* ./'
alias ss="sed -i -e 's/ //g' -e 's/dna:.\{1,1000\}//g' *.fa"
alias getaws="aws s3 cp --no-sign-request"
alias lw="ll | wc -l"
alias mmi="mamba install --freeze-installed"
alias mmb="mamba install --freeze-installed -c bioconda "
# 千万不要写 alias conda="mamba",血的教训!!!
###################################################################################################
########################################### starship ################################################
###################################################################################################
# run starship
eval "$(starship init zsh)"starship.toml配置文件:
# # Get editor completions based on the config schema
# # "$schema" = 'https://starship.rs/config-schema.json'
"$schema" = 'https://www.web4xiang.com/blog/article/starship/config-schema.json'
add_newline = true
format = """$time $all"""
[character]
success_symbol = "[👉👉👉➡️ ](white)"
error_symbol = "[👉👉👉➡️ ](red)"
[git_branch]
symbol = "🌱 "
truncation_length = 4
truncation_symbol = ""
# ignore_branches = ["master", "main"]
format = "[](fg:#ffffff bg:none)[ 🌱 ](fg:#282c34 bg:#ffffff)[](fg:#ffffff bg:#24c90a)[ $branch]($style)[](fg:#24c90a bg:none) "
style = "fg:#ffffff bg:#24c90a"
[git_commit]
commit_hash_length = 4
tag_symbol = "🔖 "
format = "[\\($hash\\)]($style) [\\($tag\\)]($style)"
style = "green"
[git_status]
format="[](fg:#ffffff bg:none)[ 🥶 ](fg:#282c34 bg:#ffffff)[ ](fg:#ffffff bg:#4da0ff)[$modified](fg:#282c34 bg:#4da0ff)[$untracked](fg:#282c34 bg:#4da0ff)[$staged](fg:#282c34 bg:#4da0ff)[$renamed](fg:#282c34 bg:#4da0ff)[](fg:#4da0ff bg:none)"
conflicted = ""
ahead = "🏎💨"
behind = "😰"
diverged = "😵"
up_to_date = "🥰"
untracked = "🤷"
stashed = "📦"
modified = "📝"
renamed = "👅"
deleted = "🗑️"
style = "red"
disabled = false
# [git_state]
# rebase = "REBASING"
# merge = "MERGING"
# revert = "REVERTING"
# cherry_pick = "CHERRY-PICKING"
# bisect = "BISECTING"
# am = "AM"
# am_or_rebase = "AM/REBASE"
# style = "yellow"
# format = '\([$state( $progress_current/$progress_total)]($style)\) '
# disabled = true
[directory]
format = "[](fg:#ffffff bg:none)[ 📚 ](fg:#282c34 bg:#ffffff)[](fg:#ffffff bg:#00b76d)[ $path]($style)[](fg:#00b76d bg:none) "
style = "fg:#ffffff bg:#00b76d"
truncation_length = 3
truncate_to_repo=false
home_symbol = ""
[nodejs]
symbol = "🤖 "
disabled = true
[package]
symbol = "📦 "
disabled = true
[python]
symbol = "🐍 "
[rust]
symbol = "🦀 "
[conda]
symbol = "🐳 "
[jobs]
symbol = "🎯 "
[time]
disabled = false
format="[](fg:#ffffff bg:none)[🐼🐻 ](fg:#282c34 bg:#ffffff)[](fg:#ffffff bg:#772953)[ $time]($style)[](fg:#772953 bg:none)"
time_format = "%T" #change to %R to only have HH:MM
style="fg:#ffffff bg:#772953"
# [custom.emoji]
# description = "Displays a (pseudo) random emoji for each prompt"
# command = "EMOJIS=(⛵️ 🛰 🪐 🚀 🧡 🔭 🌍 💫 ☄️ ✨ 🌠 🌌 ⭐️); EMOJI_INDEX=$(( $RANDOM % 12 + 0 )); echo ${EMOJIS[$EMOJI_INDEX]}"
# when = "1"
# shell = ["cmd /C"]
# format = "$output"一些有用的命令
- kill掉某个程序产生的所有进程,如kill掉EDTA产生的所有进程:
ps -ef | grep 'EDTA' | grep -v grep | awk '{print $2}' | xargs -r kill -9R语言
软件安装
基础
PCoA
# 输入数据的行是样品,列是特征变量
readr::read_table("./data/sanqimetagenome/data/all.kraken2.rrarefied.counts.txt") %>%
dplyr::select(contains("SRR")) %>%
t() %>%
as.data.frame() %>%
vegan::vegdist(method = "bray") %>%
ape::pcoa() -> pcoa.res
# 提取权重
pcoa.res$values %>%
as.data.frame() -> pcoa.weight
# 提取位置
pcoa.res$vectors %>%
as.data.frame() %>%
dplyr::select(1:5) %>%
magrittr::set_names(paste0("PCoa",1:ncol(.))) %>%
tibble::rownames_to_column(var = "Run") %>%
dplyr::left_join(df.sample.info) -> df.pcoa.point
# PCoA绘图
df.pcoa.point %>%
ggplot(aes(PCoa1, PCoa2, color = Type, shape = Part)) +
geom_hline(yintercept = 0, color = "#222222",
linetype = "dashed", linewidth = 0.5)+
geom_vline(xintercept = 0, color = "#222222",
linetype = "dashed", linewidth = 0.51) +
geom_point(size = 5)+
labs(x = "PCoA1 (21.64%)", y = "PCoA2 (8.36%)") +
scale_x_continuous(limit = c(-0.6, 0.2)) +
scale_y_continuous(limit = c(-0.2, 0.2)) +
scale_color_npg() +
pac4xiang::mytheme() +
theme(
panel.grid = element_blank(),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank(),
legend.title = element_blank(),
legend.position = c(0.2,0.8)
) -> p.pcoaPERMANOVA
# 输入数据的行是样品,列是特征变量
# Type*Part在df.sample.info这个数据框里面
vegan::adonis2(df.peranova.table ~ Type*Part,
data = df.sample.info,
permutations = 999,
method="bray") %>%
as.data.frame() %>%
tibble::rownames_to_column(var = "Index") %>%
writexl::write_xlsx("./data/sanqimetagenome/results/PERMANOVA.res.xlsx")资源
绘图
热图
- 修改
pheatmap分类信息注释信息:
ann_colors = list(
Sample.From = c(Cropland = ggsci::pal_npg()(2)[1],
Understory = ggsci::pal_npg()(2)[2]),
MAGs.From = c(Cropland = ggsci::pal_npg()(2)[1],
Understory = ggsci::pal_npg()(2)[2]),
Methods = c(metaWRAP = ggsci::pal_npg()(6)[3],
CONCOCT = ggsci::pal_npg()(6)[4],
MaxBin2 = ggsci::pal_npg()(6)[5],
MetaBAT2 = ggsci::pal_npg()(6)[6])
)Python
基因组
三代基因组组装(Hi-Fi)
软件安装
mamba install -c bioconda hifiasm
mamba install -c bioconda ragtag
mamba install -c bioconda buscohifiasm组装
nohup hifiasm -o 01.hifiasm/101 -t60 -l0 data/fq/101.fq > hifiasm.log 2>&1 &结果文件:
-rw-rw-r-- 1 lixiang lixiang 164M 10月 23 16:18 101.bp.a_ctg.gfa
-rw-rw-r-- 1 lixiang lixiang 901K 10月 23 16:18 101.bp.a_ctg.lowQ.bed
-rw-rw-r-- 1 lixiang lixiang 2.2M 10月 23 16:18 101.bp.a_ctg.noseq.gfa
-rw-rw-r-- 1 lixiang lixiang 410M 10月 23 16:17 101.bp.p_ctg.gfa
-rw-rw-r-- 1 lixiang lixiang 958K 10月 23 16:18 101.bp.p_ctg.lowQ.bed
-rw-rw-r-- 1 lixiang lixiang 11M 10月 23 16:17 101.bp.p_ctg.noseq.gfa
-rw-rw-r-- 1 lixiang lixiang 709M 10月 23 16:17 101.bp.p_utg.gfa
-rw-rw-r-- 1 lixiang lixiang 2.6M 10月 23 16:17 101.bp.p_utg.lowQ.bed
-rw-rw-r-- 1 lixiang lixiang 15M 10月 23 16:17 101.bp.p_utg.noseq.gfa
-rw-rw-r-- 1 lixiang lixiang 727M 10月 23 16:17 101.bp.r_utg.gfa
-rw-rw-r-- 1 lixiang lixiang 2.7M 10月 23 16:17 101.bp.r_utg.lowQ.bed
-rw-rw-r-- 1 lixiang lixiang 16M 10月 23 16:17 101.bp.r_utg.noseq.gfa
-rw-rw-r-- 1 lixiang lixiang 3.0G 10月 23 16:15 101.ec.bin
-rw-rw-r-- 1 lixiang lixiang 149M 10月 23 16:16 101.ovlp.reverse.bin
-rw-rw-r-- 1 lixiang lixiang 1.7G 10月 23 16:16 101.ovlp.source.bin
-rw-rw-r-- 1 lixiang lixiang 406M 10月 23 17:01 101.primary.assembly.fa
-rw-rw-r-- 1 lixiang lixiang 15K 10月 23 17:13 101.primary.assembly.fa.fai
drwxrwxr-x 2 lixiang lixiang 4.0K 10月 23 17:34 101.rag.tag其中``p_ctg.gfa是主要的contigs`.
将组装得到的结果转换为fasta格式:
gfatools gfa2fa 01.hifiasm/101.bp.p_ctg.gfa > 01.hifiasm/process/101.primary.assembly.fa挂载到染色体得到伪染色体:
ragtag.py scaffold -t 60 ~/project/yyt.3rd.acuce/02.r498/01.r498.genome/R498.genome 01.hifiasm/process/101.primary.assembly.fa -o 01.hifiasm/process/101.rag.tag提取染色体序列:
seqkit grep -f chr.id.txt 01.hifiasm/process/101.rag.tag/ragtag.scaffold.fasta > 01.hifiasm/final/101.HiFiasm.faBUSCO验证组装结果:
busco -i 01.hifiasm/final/101.HiFiasm.fa -l ~/lixiang/database/busco.plant/embryophyta_odb10 --cpu 75 -f --offline -m genome -o 01.hifiasm/busco/101---------------------------------------------------
|Results from dataset embryophyta_odb10 |
---------------------------------------------------
|C:98.7%[S:96.3%,D:2.4%],F:0.9%,M:0.4%,n:1614 |
|1593 Complete BUSCOs (C) |
|1554 Complete and single-copy BUSCOs (S) |
|39 Complete and duplicated BUSCOs (D) |
|14 Fragmented BUSCOs (F) |
|7 Missing BUSCOs (M) |
|1614 Total BUSCO groups searched |
---------------------------------------------------pbsv鉴定结构变异
# pbmm2进行比对
pbmm2 align --sort -J 60 03.mapping/01.index/r498.mmi data/raw/101/r84071_230824_001/A01/m84071_230824_103359_s1.hifi_reads.bc2027.bam -j 60 03.mapping/02.bam/101.bam# 鉴定结构变异信号
pbsv discover 03.mapping/02.bam/101.bam 05.pbsv/101.svsig.gz# 鉴定结构变异-单个样本
pbsv call -j 60 -S 0 data/ref/r498.fa 05.pbsv/101.svsig.gz 05.pbsv/101.pbsv.vcf# 鉴定结构变异-多个样本
pbsv call ref.fa ref.sample1.svsig.gz ref.sample2.svsig.gz ref.var.vcfsniffles2结构变异
这个软件的速度非常快。参考文献:
sniffles -t 60 --input 03.mapping/02.bam/1.bam --snf 08.sniffles/1.snf
sniffles -t 60 --input 08.sniffles/*.snf --vcf 08.sniffles/all.sniffles.vcf下一步是利用SV推断群体结构,这里选择不过滤位点信息,如果是后续做GWAS的话再过来位点。
将vcf文件转为bed文件:
vcftools --vcf 08.sniffles/all.sniffles.vcf --plink --out 08.sniffles/all.sniffles.for.plink.pca.vcf
plink --noweb --file 08.sniffles/all.sniffles.for.plink.pca.vcf --make-bed --out 08.sniffles/all.sniffles.for.plink.pca.vcf.final输出的文件有这些:
all.sniffles.for.plink.pca.vcf.final.bed
all.sniffles.for.plink.pca.vcf.final.bim
all.sniffles.for.plink.pca.vcf.final.fam
all.sniffles.for.plink.pca.vcf.final.log
all.sniffles.for.plink.pca.vcf.final.nosex
all.sniffles.for.plink.pca.vcf.log
all.sniffles.for.plink.pca.vcf.map
all.sniffles.for.plink.pca.vcf.ped
all.sniffles.vcf计算PCA:
plink --threads 60 --bfile 08.sniffles/all.sniffles.for.plink.pca.vcf.final --pca 3 --out 08.sniffles/all.sniffles.plink.pca.res输出的文件:
-rw-rw-r-- 1 lixiang lixiang 24 10月 29 13:04 08.sniffles/all.sniffles.plink.pca.res.eigenval
-rw-rw-r-- 1 lixiang lixiang 3.6K 10月 29 13:04 08.sniffles/all.sniffles.plink.pca.res.eigenvec
-rw-rw-r-- 1 lixiang lixiang 1.1K 10月 29 13:04 08.sniffles/all.sniffles.plink.pca.res.log
-rw-rw-r-- 1 lixiang lixiang 582 10月 29 13:04 08.sniffles/all.sniffles.plink.pca.res.nosexGFF文件注释VCF文件
使用的是vcfanno,参考文献:
先对GFF文件进行处理。
sort -k1,1 -k4,4n r498.gff > r498.sorted.gff
bgzip r498.sorted.gff
tabix r498.sorted.gff.gz两个配置文件:
gff_sv.lua:
function gff_to_gene_name(infos, name_field)
if infos == nil or #infos == 0 then
return nil
end
local result = {}
for i=1,#infos do
info = infos[i]
local s, e = string.find(info, name_field .. "=")
if s ~= nil then
name = info:sub(e + 1)
s, e = string.find(name, ";")
if e == nil then
e = name:len()
else
e = e - 1
end
result[name:sub(0, e)] = 1
end
end
local keys = {}
for k, v in pairs(result) do
keys[#keys+1] = k
end
return table.concat(keys,",")
endgff_sv.conf:
[[annotation]]
file="r498.sorted.gff.gz"
columns=[9]
names=["gene"]
ops=["lua:gff_to_gene_name(vals, 'gene_name')"]运行:
vcfanno -p 60 -lua gff_sv.lua gff_sv.conf all.sniffles.vcf > all.sniffles.vcfanno.vcf DNA甲基化
由于PacBio HiFi 的数据能够直接对DNA甲基化进行鉴定,因此加一步DNA甲基化:
~/software/pb-CpG-tools-v2.3.2/bin/aligned_bam_to_cpg_scores --bam 03.mapping/02.bam/101.bam --output-prefix ./04.CpG.methylation/101 --model ~/software/pb-CpG-tools-v2.3.2/models/pileup_calling_model.v1.tflite --threads 60重复序列注释
使用EDTA对重复序列进行注释,参考文献:
[Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline](Ou S, Su W, Liao Y, et al. Benchmarking transposable element annotation methods for creation of a streamlined, comprehensive pipeline[J]. Genome biology, 2019, 20(1): 1-18.)
软件安装
我直接用conda安装的时候失败了,选择按照官网建议的方式安装:
git clone https://github.com/oushujun/EDTA.git
conda env create -f EDTA.yml测试是否安装成功:
cd ./EDTA/test
EDTA.pl --genome genome.fa --cds genome.cds.fa --curatedlib ../database/rice6.9.5.liban --exclude genome.exclude.bed --overwrite 1 --sensitive 1 --anno 1 --evaluate 1 --threads 10输入文件
基因组文件是必须的,其他可选的文件有:
- 编码序列:可以是这个物种的,也可以是近缘种的;
- 基因组上基因的起始和终止位置;
- 精准的物种TE库,如果没有就最好不要加进去。
运行示例
EDTA.pl --species Rice --genome ../02.hifiasm/final/1.HiFiasm.fa --cds ../01.data/ref/r498.cds.fa --curatedlib ../01.data/ref/r498.te.bed --overwrite 1 --sensitive 1 --evaluate 1 --threads 60Augustus基因注释
augustus --species=rice --protein=on --codingseq=on --introns=on --start=on --stop=on --cds=on --exonnames=on --gff3=on 02.hifiasm/final/1.HiFiasm.fa > 14.augustus/1.augustus.gff3DeepVariant鉴定变异
安装
直接使用docker安装:
docker pull google/deepvariant:1.6.0数据准备
需要bam文件和基因组文件。bam问价可以使用minimap比对得到,需要使用samtools构建索引,生成.bai文件;基因组文件需要使用samtools faidx构建索引。
开始运行
我尝试使用基因组间的比对结果进行变异检测,但是失败了,换成原始数据比对得到的bam文件就可以了:
docker run \
-v "/home/lixiang/lixiang/yyt.acuce.3rd/03.mapping/02.bam/":"/input" \
-v "/home/lixiang/lixiang/yyt.acuce.3rd/19.deepvariant/":"/output" \
google/deepvariant:1.6.0 /opt/deepvariant/bin/run_deepvariant \
--model_type=WGS \
--ref=/input/r498.chr1-ch12.fa \
--reads=/input/99.bam \
--output_vcf=/output/vcf/99.vcf.gz \
--output_gvcf=/output/gvcf/99.gvcf.gz \
--num_shards=50 \
--logging_dir=/output/log/logs \
--dry_run=false \
--sample_name 99一行代码:
docker run -v "/home/lixiang/lixiang/yyt.acuce.3rd/03.mapping/02.bam/":"/input" -v "/home/lixiang/lixiang/yyt.acuce.3rd/19.deepvariant/":"/output" google/deepvariant:1.6.0 /opt/deepvariant/bin/run_deepvariant --model_type=WGS --ref=/input/r498.chr1-ch12.fa --reads=/input/99.bam --output_vcf=/output/vcf/99.vcf.gz --output_gvcf=/output/gvcf/99.gvcf.gz --num_shards=50 --logging_dir=/output/log/logs --dry_run=false --sample_name 99变异数量统计
用bedtools统计染色体不同区域上变异的覆盖度。
# 构建基因组索引
samtools faidx r498.chr1-ch12.fa
# 基因组划窗
bedtools makewindows -g r498.chr1-ch12.fa.fai -w 100000 > r498.genome.window.100000.bed
# 计算覆盖度
bedtools coverage -a r498.genome.window.100000.bed -b vcf/1.vcf.gz -counts > snp.coverage/1.variant.deepvariant.coverage.txtSyRi鉴定结构变异
对结构变异进行注释:
- 提取结构变异的位置信息形成
bed文件:
awk '$4 !="-"' ../15.syri/02.sryi/1.HiFiasm.syrisyri.out | cut -f 1-3,9 | > ./1.HiFiasm.syrisyri.bed#染色体 起始位置 终止位置 变异编号
Chr1 9522 9522 SNP3865
Chr1 9523 9523 SNP3866
Chr1 9570 9571 DEL3867
Chr1 9608 9608 INS3868
Chr1 9674 9674 INS3869
Chr1 9717 9717 SNP3870
Chr1 9757 9757 INS3871
Chr1 9794 9795 DEL3872
Chr1 9813 9815 DEL3873
Chr1 9822 9823 DEL3874- 提取
gff文件中gene的信息,形成bed文件:
awk '$3 =="gene"' r498.gff | awk {'print $1,$4, $5, $9'} | awk '{split($4, arr, ";"); print $1, $2, $3, arr[1]}' | sed 's/ /\t/g' | sed 's/ID=//g' > r498.gene.bed# 染色体 起始位置 终止位置 基因ID
Chr10 2763040 2763714 OsR498G1018107300.01
Chr10 20368540 20372915 OsR498G1018933700.01
Chr10 1287616 1287972 OsR498G1069497000.01
Chr10 4105970 4107094 OsR498G1069588100.01
Chr10 7942000 7943565 OsR498G1018367300.01
Chr10 13557765 13562571 OsR498G1018574200.01
Chr10 19655553 19661142 OsR498G1018894000.01
Chr10 19144811 19147243 OsR498G1018866300.01
Chr10 23208493 23210268 OsR498G1019111000.01
Chr10 3002409 3003778 OsR498G1018118600.01- 合并:
bedtools intersect -a 1.HiFiasm.syrisyri.bed -b r498.gene.bed -loj -wo > 1.HiFiasm.syrisyri.sv.gene.txt 没有和基因位置有交集的会用占位符占位:
Chr1 466785 466785 SNP4173 . -1 -1 .
Chr1 468345 468345 SNP4174 . -1 -1 .
Chr1 472130 472133 DEL4175 . -1 -1 .
Chr1 484574 484574 SNP4176 Chr1 482644 485461 OsR498G0100029700.01
Chr1 484578 484583 DEL4177 Chr1 482644 485461 OsR498G0100029700.01
Chr1 485537 485537 SNP4178 . -1 -1 .
Chr1 486206 486206 INS4179 . -1 -1 .
Chr1 486289 486289 INS4180 . -1 -1 .
Chr1 486673 486673 SNP4181 Chr1 486574 487163 OsR498G0100029900.01
Chr1 486677 486677 SNP4182 Chr1 486574 487163 OsR498G0100029900.01
Chr1 487347 487347 SNP4183 . -1 -1 .如果只想要注释到基因上的变异位点,使用:
bedtools intersect -a 1.HiFiasm.syrisyri.bed -b r498.gene.bed -wa -wb > 1.HiFiasm.syrisyri.sv.gene.txt输出的结果就全是注释到基因的变异位点:
Chr1 32047 32047 SNP3883 Chr1 24850 34201 OsR498G0100001100.01
Chr1 47307 47315 DEL3884 Chr1 46834 50926 OsR498G0100002100.01
Chr1 51885 51885 INS3885 Chr1 51148 52666 OsR498G0100002300.01
Chr1 229993 229993 SNP3900 Chr1 226316 230889 OsR498G0100015500.01
Chr1 281669 281669 SNP3908 Chr1 278278 285269 OsR498G0100017900.01
Chr1 302175 302175 INS3910 Chr1 296187 309155 OsR498G0100018900.01
Chr1 302551 302551 SNP3911 Chr1 296187 309155 OsR498G0100018900.01
Chr1 307894 307894 SNP3912 Chr1 296187 309155 OsR498G0100018900.01
Chr1 307942 307942 INS3913 Chr1 296187 309155 OsR498G0100018900.01
Chr1 322957 322958 DEL3918 Chr1 321670 324907 OsR498G0100021200.01VCF文件建树
大概可以分为过滤、计算距离、建树等三步。
- 过滤低质量的变异位点:
bcftools filter -i 'QUAL>=20' all.deepvariant.vcf.gz -O z -o all.deepvariant.filtered.vcf.gz- 筛选SNP数据:
bcftools view -v snps all.deepvariant.filtered.vcf.gz -Oz -o all.deepvariant.filtered.snp.vcf.gz- 再次过滤SNP:
plink --vcf all.deepvariant.filtered.cnp.vcf.gz --geno 0.1 --maf 0.05 --out all.deepvariant.filtered.snp.plink --recode vcf-iid --allow-extra-chr --set-missing-var-ids @:# --keep-allele-order- 转换为phylip格式:
python3 ~/software/vcf2phylip-2.8/vcf2phylip.py -i all.deepvariant.filtered.snp.plink.vcf- IQ-tree建树:
-m MFP表示查找最佳模型,-b表示Bootstrap 迭代次数。为了节省时间,看个大概,可以使用-m TEST。
iqtree --mem 80% -T 60 -m MFP -b 1000 -s all.deepvariant.filtered.snp.plink.min4.phy
-m MFP:ModelFinder will test up to 1232 protein models (sample size: 168525) …
-m TEST:ModelFinder will test up to 224 protein models (sample size: 168525) …
运行完成会输出最佳模型:
Best-fit model: PMB+F+I+G4 chosen according to BIC.
- 计算距离
~/software/VCF2Dis-1.50/bin/VCF2Dis -i all.deepvariant.filtered.snp.vcf.gz -o all.deepvariant.filtered.snp.mat - 构建nj-tree
````
### VCF文件PCA
- 过滤低质量的变异位点:
````sh
bcftools filter -i 'QUAL>=20' all.deepvariant.vcf.gz -O z -o all.deepvariant.filtered.vcf.gz- 筛选SNP数据:
bcftools view -v snps all.deepvariant.filtered.vcf.gz -Oz -o all.deepvariant.filtered.snp.vcf.gz- 再次过滤SNP:
plink --vcf all.deepvariant.filtered.cnp.vcf.gz --geno 0.1 --maf 0.05 --out all.deepvariant.filtered.snp.plink --recode vcf-iid --allow-extra-chr --set-missing-var-ids @:# --keep-allele-order- 格式转换:将vcf文件转换为plink格式。
vcftools --vcf all.deepvariant.filtered.snp.plink.vcf --plink --out all.deepvariant.filtered.snp.plink.pca.data
plink --noweb --file all.deepvariant.filtered.snp.plink.pca.data --make-bed --out all.deepvariant.filtered.snp.plink.pca.data.final- PCA
plink --bfile all.deepvariant.filtered.snp.plink.pca.data.final --pca --out all.deepvariant.filtered.snp.plink.pca.result最终的文件有这些:
-rw-rw-r-- 1 lixiang lixiang 308 11月 14 19:54 all.deepvariant.filtered.snp.plink.nosex
-rw-rw-r-- 1 lixiang lixiang 2.3M 11月 14 20:21 all.deepvariant.filtered.snp.plink.pca.data.final.bed
-rw-rw-r-- 1 lixiang lixiang 5.1M 11月 14 20:21 all.deepvariant.filtered.snp.plink.pca.data.final.bim
-rw-rw-r-- 1 lixiang lixiang 794 11月 14 20:21 all.deepvariant.filtered.snp.plink.pca.data.final.fam
-rw-rw-r-- 1 lixiang lixiang 1.6K 11月 14 20:21 all.deepvariant.filtered.snp.plink.pca.data.final.log
-rw-rw-r-- 1 lixiang lixiang 308 11月 14 20:21 all.deepvariant.filtered.snp.plink.pca.data.final.nosex
-rw-rw-r-- 1 lixiang lixiang 532 11月 14 20:19 all.deepvariant.filtered.snp.plink.pca.data.log
-rw-rw-r-- 1 lixiang lixiang 4.4M 11月 14 20:19 all.deepvariant.filtered.snp.plink.pca.data.map
-rw-rw-r-- 1 lixiang lixiang 36M 11月 14 20:19 all.deepvariant.filtered.snp.plink.pca.data.ped
-rw-rw-r-- 1 lixiang lixiang 166 11月 14 20:22 all.deepvariant.filtered.snp.plink.pca.result.eigenval
-rw-rw-r-- 1 lixiang lixiang 12K 11月 14 20:22 all.deepvariant.filtered.snp.plink.pca.result.eigenvec
-rw-rw-r-- 1 lixiang lixiang 1.1K 11月 14 20:22 all.deepvariant.filtered.snp.plink.pca.result.log
-rw-rw-r-- 1 lixiang lixiang 308 11月 14 20:22 all.deepvariant.filtered.snp.plink.pca.result.nosex
-rw-rw-r-- 1 lixiang lixiang 42M 11月 14 19:54 all.deepvariant.filtered.snp.plink.vcf
-rw-rw-r-- 1 lixiang lixiang 725M 11月 14 18:36 all.deepvariant.filtered.vcf.gz
-rw-rw-r-- 1 lixiang lixiang 8.3G 11月 14 17:23 all.deepvariant.vcf.gz用all.deepvariant.filtered.snp.plink.pca.result.eigenval和all.deepvariant.filtered.snp.plink.pca.result.eigenvec这两个文件就可以画图了。
ANNOVAR注释变异
- gff3文件转GenPred文件(PS:GFF3文件需要有表头,加上
##gff-version 3即可。)
gff3ToGenePred KY131.IGDBv1.Allset.gff KY131_refGene.txt- 提取转录本序列
perl ~/software/annovar/retrieve_seq_from_fasta.pl --format refGene --seqfile KY131.genome KY131_refGene.txt -outfile KY131_refGeneMrna.fa - 过滤并转换VCF格式
bcftools filter -i 'QUAL>=20' 07.gatk/s-1-2_H7GYJCCXY_L1.RGA5.gvcf -O z -o 07.gatk/s-1-2_H7GYJCCXY_L1.RGA5.gvcf.filtered.gz
perl ~/software/annovar/convert2annovar.pl -format vcf4 07.gatk/s-1-2_H7GYJCCXY_L1.RGA5.gvcf.filtered.gz > 07.gatk/s-1-2_H7GYJCCXY_L1.RGA5.annovar.vcf- 注释
perl ~/software/annovar/annotate_variation.pl --geneanno -dbtype refGene --buildver KY131 07.gatk/s-1-2_H7GYJCCXY_L1.RGA5.annovar.vcf 01.data/ANNOVAR -out 10.annovar/s-1-2_H7GYJCCXY_L1.RGA5输出结果:每个文件会输出三个文件
-rw-rw-r-- 1 lixiang lixiang 36K 11月 24 15:39 s-101_H7YKKCCXY_L1.PIK1.exonic_variant_function
-rw-rw-r-- 1 lixiang lixiang 1.1K 11月 24 15:39 s-101_H7YKKCCXY_L1.PIK1.log
-rw-rw-r-- 1 lixiang lixiang 31K 11月 24 15:39 s-101_H7YKKCCXY_L1.PIK1.variant_function- s-101_H7YKKCCXY_L1.PIK1.exonic_variant_function:变异信息,是同义突变还是非同义突变,以及突变前后的序列差异。
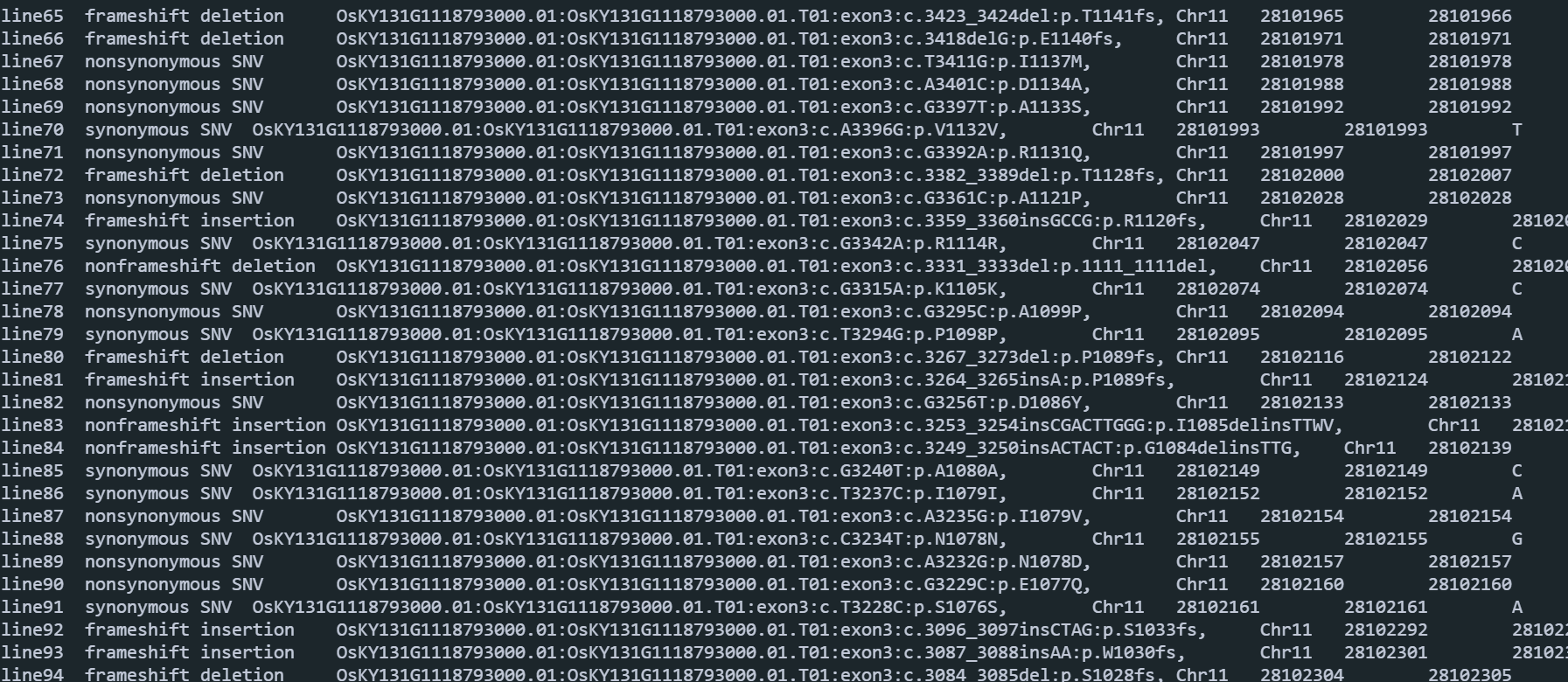
- s-101_H7YKKCCXY_L1.PIK1.variant_function:突变发生的位置在那个区域。
转录组
可变剪切分析
参考文献
软件安装
mamba install -c bioconda rmats构建索引
STAR --runMode genomeGenerate --genomeDir 01.genome.data/star.index --genomeFastaFiles 01.genome.data/genome.fa --sjdbGTFfile 01.genome.data/genome.gtf泛基因组
graph-based pan-genome
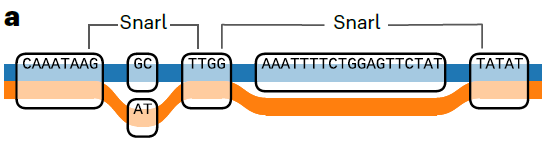
如何理解基于图的泛基因组呢?可以想象成一个网络图,有节点node和边edge,节点就是没有发生变异的序列,edge呢就是发生了遗传变异的序列,将这些节点连接起来就得到path.
Minigraph-Cactus
这个流程发表在NBT上(Pangenome graph construction from genome alignments with Minigraph-Cactus),人类泛基因组文章用的就是这个流程。
软件安装
软甲地址是https://github.com/ComparativeGenomicsToolkit/cactus. 自己编译,但是我编译好几次都没有泛基因组这个流程,索性用Docker直接部署。
docker pull quay.io/comparative-genomics-toolkit/cactus:v2.7.0软件使用
- 样品文件准备:准备一个txt文件,以
\t分隔,第一列是样品名称,第二列是样品所在的路径:
100 ./genome/100.HiFiasm.fa
101 ./genome/101.HiFiasm.fa
10 ./genome/10.HiFiasm.fa
11 ./genome/11.HiFiasm.fa
12 ./genome/12.HiFiasm.fa
13 ./genome/13.HiFiasm.fa
14 ./genome/14.HiFiasm.fa
15 ./genome/15.HiFiasm.fa
16 ./genome/16.HiFiasm.fa
17 ./genome/17.HiFiasm.fa- 进入镜像:需要指定宿主机的工作目录,也就是文件存放的位置,如我的工作路径在
~/lixiang/yyt.acuce.3rd/25.pangenome:
docker run -itd --name cactus-pan -v ~/lixiang/yyt.acuce.3rd/25.pangenome:/data quay.io/comparative-genomics-toolkit/cactus:v2.7.0- 开始运行:100个水稻的基因组差不多24小时就能运行完了,速度还是很快的:
nohup docker exec cactus-pan cactus-pangenome ./jb sample.txt --outDir ./cactus-pangenome.r498 --outName Acuce --reference R498 --vcf --giraffe --gfa --gbz --odgi --xg --viz --draw --chrom-vg --chrom-og --maxMemory 400G --logFile run.log - 输出文件:输出文件包含了最常见的
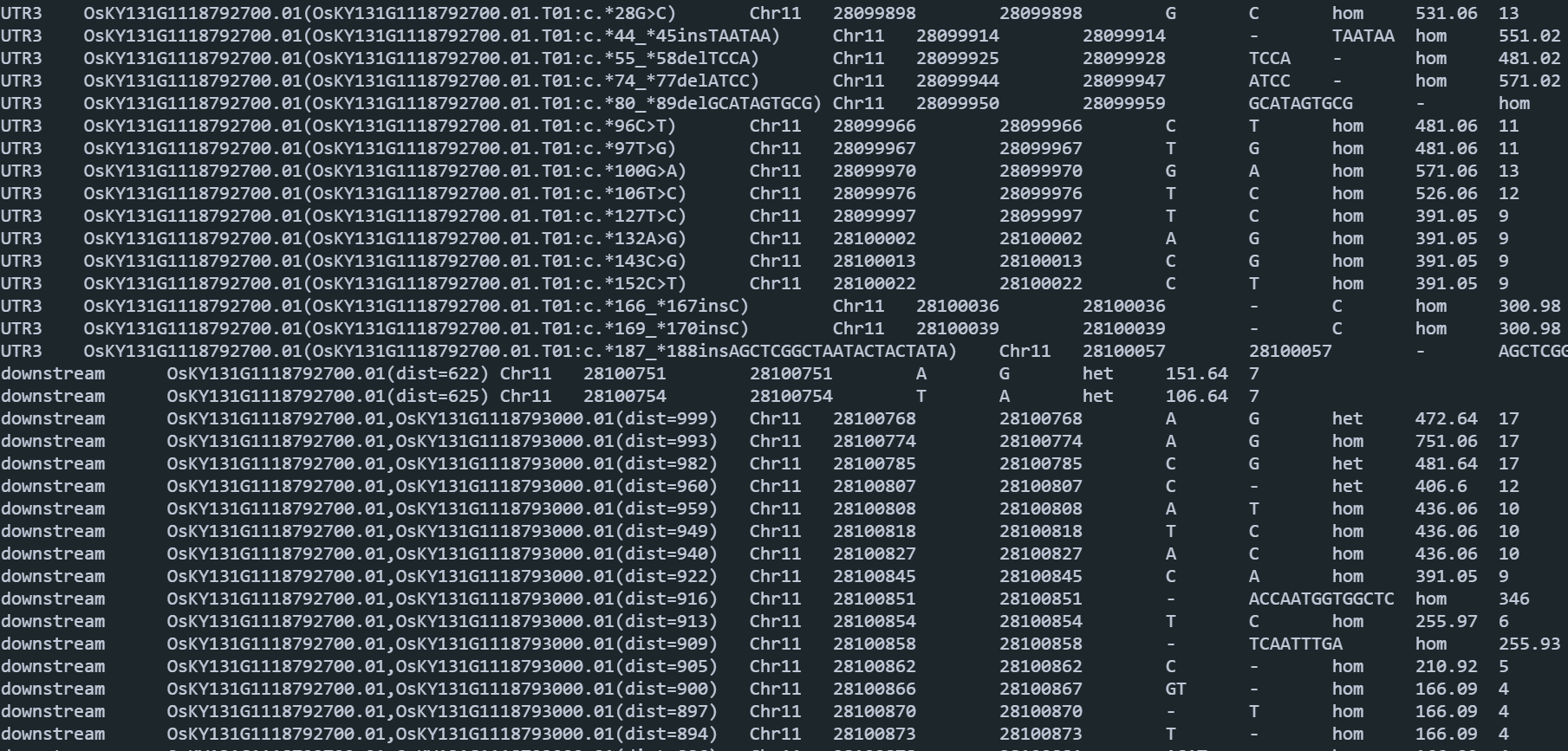
gfa格式的graph文件,vcf文件存放变异信息,chrom-subproblems文件夹包含了每条染色体的graph。
总计 38G
drwxr-xr-x 2 root root 4.0K 12月 24 02:33 Acuce.chroms
-rw-r--r-- 1 root root 323M 12月 24 02:33 Acuce.d2.dist
-rw-r--r-- 1 root root 366M 12月 24 02:33 Acuce.d2.gbz
-rw-r--r-- 1 root root 4.8G 12月 24 02:33 Acuce.d2.min
-rw-r--r-- 1 root root 13G 12月 24 02:33 Acuce.full.hal
-rw-r--r-- 1 root root 6.6G 12月 24 02:33 Acuce.full.og
-rw-r--r-- 1 root root 316M 12月 23 14:26 Acuce.gaf.gz
-rw-r--r-- 1 root root 367M 12月 24 02:33 Acuce.gbz
-rw-r--r-- 1 root root 126M 12月 23 14:27 Acuce.gfa.fa.gz
-rw-r--r-- 1 root root 1015M 12月 24 02:33 Acuce.gfa.gz
-rw-r--r-- 1 root root 1.8G 12月 23 14:26 Acuce.paf
-rw-r--r-- 1 root root 1.7M 12月 23 14:26 Acuce.paf.filter.log
-rw-r--r-- 1 root root 440M 12月 23 14:26 Acuce.paf.unfiltered.gz
-rw-r--r-- 1 root root 1.3G 12月 24 02:33 Acuce.raw.vcf.gz
-rw-r--r-- 1 root root 186K 12月 24 02:33 Acuce.raw.vcf.gz.tbi
-rw-r--r-- 1 root root 17M 12月 24 02:33 Acuce.snarls
-rw-r--r-- 1 root root 477K 12月 24 02:33 Acuce.stats.tgz
-rw-r--r-- 1 root root 122M 12月 23 10:03 Acuce.sv.gfa.gz
-rw-r--r-- 1 lixiang lixiang 2.1G 12月 24 02:33 Acuce.vcf
-rw-r--r-- 1 root root 179K 12月 24 02:33 Acuce.vcf.gz.tbi
drwxr-xr-x 2 root root 4.0K 12月 24 02:33 Acuce.viz
-rw-r--r-- 1 root root 5.9G 12月 24 02:33 Acuce.xg
drwxr-xr-x 2 root root 4.0K 12月 23 18:40 chrom-alignments
drwxr-xr-x 18 root root 4.0K 12月 23 14:33 chrom-subproblems
-rw-r--r-- 1 root root 3.7K 12月 23 14:27 sample.txt比较基因组
软件安装
使用mamba安装软件:
mamba create --name comparative.genomics
mamba activate comparative.genomics
# blast+
mamba install -c bioconda blast
# OrthoFinder
mmaba install -c bioconda orthofinder
# gffread=0.99
# 脚本需要这个版本
mamba install -c bioconda gffread=0.9.9
# bio-per
# 脚本需要用到这个perl模块
mamba install -c bioconda perl-bioperl
mamba install -c bioconda gblocks
mamba install -c bioconda pal2nal
mamba install -c bioconda paml
mamba install -c bioconda cafe需要手动安装FigTree,主要功能是将系统发育树设置为有根树。
官方地址:https://github.com/rambaut/figtree/releases/tag/v1.4.4.
点击下载Windows版本,解压后双击打开即可。
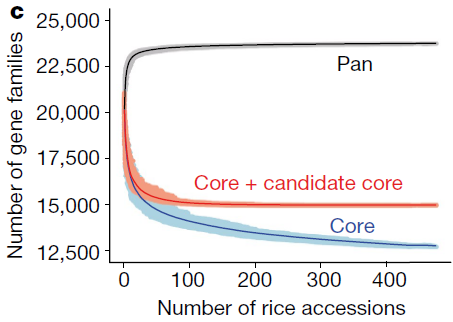
数据下载处理
从Rice Resource Center下载非洲栽培稻CG14、籼稻代表性品种IR64、R498和我们国内水稻研究领域常用的ZH11用于本次分析。最终得到如下的文件:
-rw-r--r-- 1 lixiang lixiang 338M 12月 19 2019 CG14.fa
-rw-r--r-- 1 lixiang lixiang 103M 12月 25 2019 CG14.gff3
-rw-rw-r-- 1 lixiang lixiang 374M 8月 8 16:38 IR64.fa
-rw-rw-r-- 1 lixiang lixiang 159M 8月 8 16:37 IR64.gff3
-rw-r--r-- 1 lixiang lixiang 364M 12月 19 2019 ZH11.fa
-rw-r--r-- 1 lixiang lixiang 135M 12月 25 2019 ZH11.gff3一个比较重要的点:基因组中染色体的编号和gff文件中的必须一致!!!如:
>grep ">" CG14.fa| head
>Chr1
>Chr2
>Chr3
>Chr4
>Chr5
>Chr6
>Chr7
>Chr8
>Chr9
>Chr10>head CG14.gff3
Chr1 IGDBv1 gene 1264 1617 . - . ID=OsCG14G0100000200.01;Name=OsCG14G0100000200.01;
Chr1 IGDBv1 mRNA 1264 1617 . - . ID=OsCG14G0100000200.01.T01;Name=OsCG14G0100000200.01.T01;Parent=OsCG14G0100000200.01;
Chr1 IGDBv1 exon 1597 1617 . - . ID=exon239804;Name=exon239804;Parent=OsCG14G0100000200.01.T01
Chr1 IGDBv1 exon 1458 1594 . - . ID=exon239805;Name=exon239805;Parent=OsCG14G0100000200.01.T01
Chr1 IGDBv1 exon 1264 1384 . - . ID=exon239806;Name=exon239806;Parent=OsCG14G0100000200.01.T01
Chr1 IGDBv1 CDS 1597 1617 . - 0 ID=cds239804;Name=cds239804;Parent=OsCG14G0100000200.01.T01
Chr1 IGDBv1 CDS 1458 1594 . - 0 ID=cds239805;Name=cds239805;Parent=OsCG14G0100000200.01.T01
Chr1 IGDBv1 CDS 1264 1384 . - 2 ID=cds239806;Name=cds239806;Parent=OsCG14G0100000200.01.T01
Chr1 IGDBv1 stop_codon 1264 1266 . - . Parent=OsCG14G0100000200.01.T01
Chr1 IGDBv1 gene 3872 4408 . - . ID=OsCG14G0100000600.01;Name=OsCG14G0100000600.01;下一步是准备CDS文件和蛋白序列文件。使用这两个脚本:get_cds_pep_from_gff_and_genome.pl、get_longest_pepV2.pl和accordingIDgetGFF.pl
使用方法为:
perl ./01.scripts/get_cds_pep_from_gff_and_genome.pl -gff ./02.data/CG14.gff3 -genome ./02.data/CG14.fa -index CG14 -od ./03.cds.pep/CG14
perl ./01.scripts/get_cds_pep_from_gff_and_genome.pl -gff ./02.data/ZH11.gff3 -genome ./02.data/ZH11.fa -index ZH11 -od ./03.cds.pep/ZH11
perl ./01.scripts/get_cds_pep_from_gff_and_genome.pl -gff ./02.data/IR64.gff3 -genome ./02.data/IR64.fa -index IR64 -od ./03.cds.pep/IR64
perl ./01.scripts/get_cds_pep_from_gff_and_genome.pl -gff ./02.data/R498.gff3 -genome ./02.data/R498.fa -index R498 -od ./03.cds.pep/R498运行完成就可以得到下面的结果:
>ll 03.cds.pep/CG14/Longest
总计 89M
-rw-rw-r-- 1 lixiang lixiang 1.6M 8月 8 16:00 CG14.gff_for_MCSCANX.txt
-rw-rw-r-- 1 lixiang lixiang 1.8M 8月 8 16:00 CG14.gff_for_WGD.txt
-rw-rw-r-- 1 lixiang lixiang 1.7M 8月 8 16:00 CG14.id
-rw-rw-r-- 1 lixiang lixiang 41M 8月 8 16:00 CG14.longest.cds.fa
-rw-rw-r-- 1 lixiang lixiang 30M 8月 8 16:0基因家族推断
使用OrthoFinder. 需要注意的点有:
- 输入文件是蛋白序列,以
.fa或者.fasta结尾; - 不能存在可变剪切,输入每个基因最长的转录本;
- 多倍体必须拆成亚基因组对应的蛋白质序列去做。如陆地棉为
AADD,共获得两套亚基因组A和D,所以要把该基因组所有的蛋白分成A和D这两个物种去做。
拷贝最长转录本的蛋白序列
mkdir 04.longest.pep
cp 03.cds.pep/CG14/Longest/CG14.longest.protein.fa 04.longest.pep/CG14.fa
cp 03.cds.pep/ZH11/Longest/ZH11.longest.protein.fa 04.longest.pep/ZH11.fa
cp 03.cds.pep/IR64/Longest/IR64.longest.protein.fa 04.longest.pep/IR64.fa
cp 03.cds.pep/R498/Longest/R498.longest.protein.fa 04.longest.pep/R498.fa运行OrthoFinder
orthofinder -f 04.longest.pep -S diamond -t 50 -n 05.orthofinder
mkdir 05.orthofinder
mv 04.longest.pep/OrthoFinder/Results_05.orthofinder/* 05.orthofinder-S表示使用什么软件进行蛋白序列比对,diamond非常快,通常用它。-t表示线程数,数字越大运行越快。
Orthofinder输出结果
运行完成会输出如下的结果:
OrthoFinder assigned 108252 genes (94.3% of total) to 31664 orthogroups. Fifty percent of all genes were in orthogroups with 3 or more genes (G50 was 3) and were contained in the largest 12883 orthogroups (O50 was 12883). There were 24518 orthogroups with all species present and 19801 of these consisted entirely of single-copy genes.
输出这些文件(主要包含共有基因家族和共有基因、各物种特有基因家族和特有基因、单拷贝基因家族和单拷贝基因和未能形成基因家族的物种特有基因):
-rw-rw-r-- 1 lixiang lixiang 2.5K 8月 8 16:50 Citation.txt
drwxrwxr-x 2 lixiang lixiang 4.0K 8月 8 16:50 Comparative_Genomics_Statistics
drwxrwxr-x 2 lixiang lixiang 4.0K 8月 8 16:50 Gene_Duplication_Events
drwxrwxr-x 2 lixiang lixiang 216K 8月 8 16:49 Gene_Trees
-rw-rw-r-- 1 lixiang lixiang 748 8月 8 16:50 Log.txt
drwxrwxr-x 2 lixiang lixiang 4.0K 8月 8 16:49 Orthogroups
drwxrwxr-x 2 lixiang lixiang 1.1M 8月 8 16:49 Orthogroup_Sequences
drwxrwxr-x 5 lixiang lixiang 4.0K 8月 8 16:50 Orthologues
drwxrwxr-x 2 lixiang lixiang 4.0K 8月 8 16:49 Phylogenetically_Misplaced_Genes
drwxrwxr-x 2 lixiang lixiang 4.0K 8月 8 16:49 Phylogenetic_Hierarchical_Orthogroups
drwxrwxr-x 2 lixiang lixiang 4.0K 8月 8 16:49 Putative_Xenologs
drwxrwxr-x 2 lixiang lixiang 216K 8月 8 16:49 Resolved_Gene_Trees
drwxrwxr-x 2 lixiang lixiang 532K 8月 8 16:49 Single_Copy_Orthologue_Sequences
drwxrwxr-x 2 lixiang lixiang 4.0K 8月 8 16:49 Species_Tree
drwxrwxr-x 5 lixiang lixiang 4.0K 8月 8 16:50 WorkingDirectory不同物种共有的Orthogroups
共有和特有的Orthogroups:
>cat 05.orthofinder/Comparative_Genomics_Statistics/Orthogroups_SpeciesOverlaps.tsv
CG14 IR64 ZH11
CG14 28526 26818 26007
IR64 26818 29829 27297
ZH11 26007 27297 28913主要统计信息
>cat 05.orthofinder/Comparative_Genomics_Statistics/Statistics_Overall.tsv
Number of species 3 #用于分析的物种数量
Number of genes 114839 #所有基因数量
Number of genes in orthogroups 108252 # 聚类成功的基因数量
Number of unassigned genes 6587 # 没有聚类上的基因数量
Percentage of genes in orthogroups 94.3 # 聚类成功的基因比例
Percentage of unassigned genes 5.7 # 未聚类的基因的比例
Number of orthogroups 31664 # 基因家族数量
Number of species-specific orthogroups 578 # 物种特异性基因家族数量
Number of genes in species-specific orthogroups 2296 # 物种特异性基因家族包含的基因数量
Percentage of genes in species-specific orthogroups 2.0 # 物种特有基因家族中包含的基因总数占比
Mean orthogroup size 3.4 # 基因家族平均基因数量
Median orthogroup size 3.0 # 基因家族基因数量的中位数
G50 (assigned genes) 3 # 按照基因家族中包含基因数目从大到小排列,排列到50%(按照聚类上基因数目统计)的基因时对应的那个基因家族包含的基因数目时对应的那个基因家族包含的基因数目
G50 (all genes) 3 # 按照全部基因统计
O50 (assigned genes) 11785 #
O50 (all genes) 12883
Number of orthogroups with all species present 24518 # 所有物种都含有的基因家族数量
Number of single-copy orthogroups 19801 # 单拷贝基因家族数量
# 下面的是其他统计信息
Date 2023-08-08
Orthogroups file Orthogroups.tsv
Unassigned genes file Orthogroups_UnassignedGenes.tsv
Per-species statistics Statistics_PerSpecies.tsv
Overall statistics Statistics_Overall.tsv
Orthogroups shared between species Orthogroups_SpeciesOverlaps.tsv
Average number of genes per-species in orthogroup Number of orthogroups Percentage of orthogroups Number of genes Percentage of genes
<1 5512 17.4 11024 10.2
'1 23452 74.1 73919 68.3
'2 1953 6.2 12442 11.5
'3 419 1.3 4015 3.7
'4 145 0.5 1828 1.7
'5 58 0.2 913 0.8
'6 40 0.1 747 0.7
'7 17 0.1 378 0.3
'8 15 0.0 375 0.3
'9 13 0.0 359 0.3
'10 6 0.0 187 0.2
11-15 19 0.1 709 0.7
16-20 9 0.0 482 0.4
21-50 4 0.0 352 0.3
51-100 2 0.0 522 0.5
101-150 0 0.0 0 0.0
151-200 0 0.0 0 0.0
201-500 0 0.0 0 0.0
501-1000 0 0.0 0 0.0
'1001+ 0 0.0 0 0.0
Number of species in orthogroup Number of orthogroups
1 578 # 578个基因存在于1个物种中
2 6568 # 6568个基因存在于2个物种中
3 24518 # 24518个基因存在于3个物种中【PS:这就是泛基因组中的core gene啊】物种统计信息
>cat 05.orthofinder/Comparative_Genomics_Statistics/Statistics_PerSpecies.tsv | 比较指标 | CG14 | IR64 | ZH11 |
|---|---|---|---|
| Number of genes【用于计算的基因数量】 | 38016 | 39498 | 37325 |
| Number of genes in orthogroups【参与聚类的基因数量】 | 35327 | 37146 | 35779 |
| Number of unassigned genes【未参与聚类的基因数量】 | 2689 | 2352 | 1546 |
| Percentage of genes in orthogroups【参与聚类的基因比例】 | 92.9 | 94 | 95.9 |
| Percentage of unassigned genes【未参与聚类的基因比例】 | 7.1 | 6 | 4.1 |
| Number of orthogroups containing species【基因家族数量】 | 28526 | 29829 | 28913 |
| Percentage of orthogroups containing species【基因家族基因比例】 | 90.1 | 94.2 | 91.3 |
| Number of species-specific orthogroups【特有的基因家族】 | 219 | 232 | 127 |
| Number of genes in species-specific orthogroups【特有的基因数量】 | 756 | 827 | 713 |
| Percentage of genes in species-specific orthogroups【特有基因的比例】 | 2 | 2.1 | 1.9 |
PS:用这个表基因可以做泛基因组分析中那个典型的图了。
结果可视化:
rm(list = ls())
library(tidyverse)
library(ggsci)
readxl::read_excel("./Statistics_PerSpecies.xlsx") %>%
tidyr::pivot_longer(cols = 3:ncol(.)) %>%
dplyr::group_by(group, name, `Copy number`) %>%
dplyr::summarise(sum = sum(value)) %>%
dplyr::ungroup() %>%
ggplot(aes(sum, name, fill = `Copy number`)) +
geom_col() +
labs(x = "Value", y = "Variety") +
facet_wrap(.~group, scales = "free") +
scale_fill_npg() +
theme_bw()
ggsave(file = "./Statistics_PerSpecies.png",
width = 8, height = 6, dpi = 500)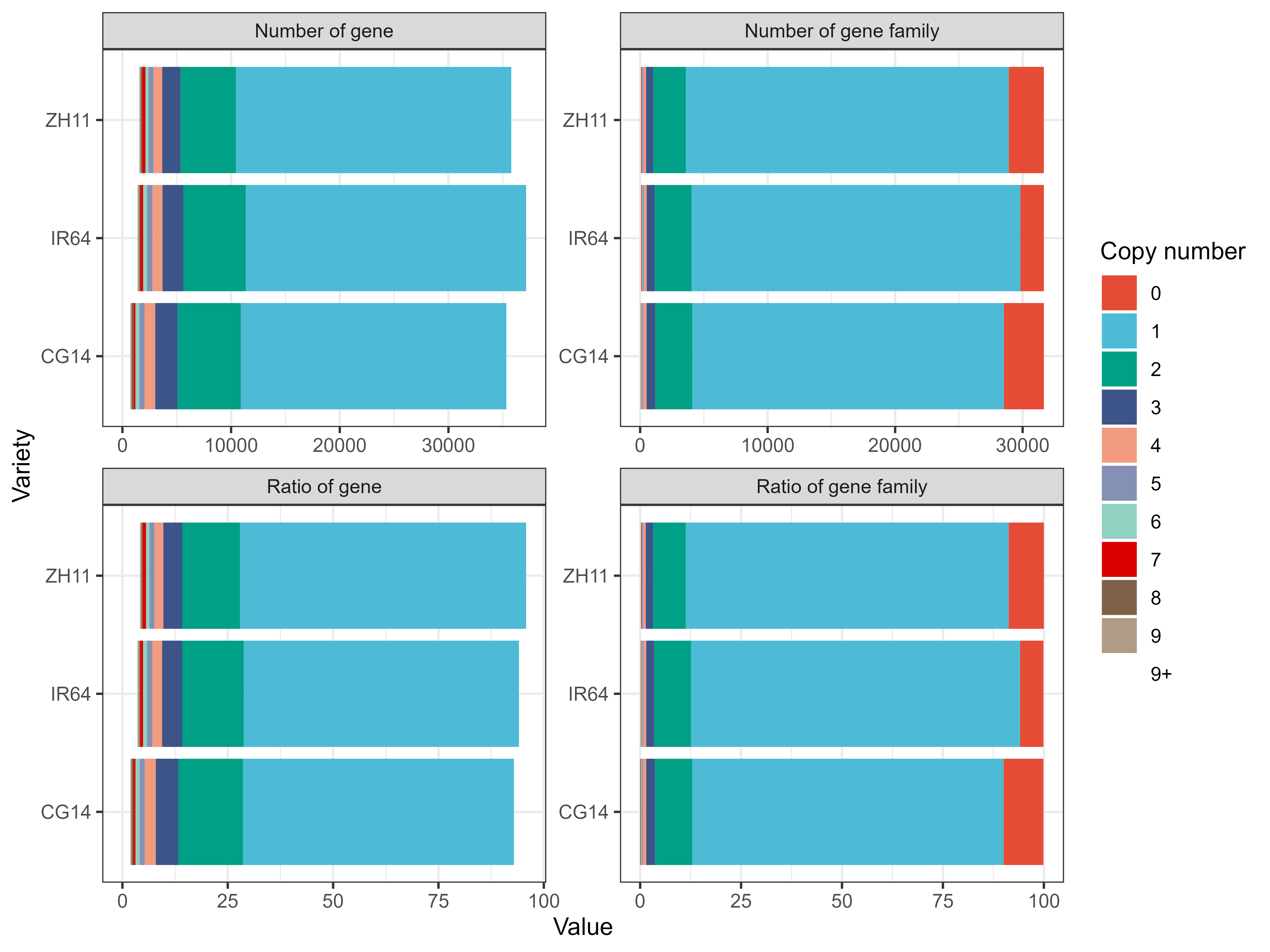
Orthogroups文件夹
Orthogroups.tsv:每一行是一个基因家族,后面的每一列是每个基因家族在每个品种中的基因编号。

Orthogroups.txt: 类似于Orthogroups.tsv,只不过是OrthoMCL的输出格式。Orthogroups_UnassignedGenes.tsv:物种特异性基因,没有聚类为基因家族的。

Orthogroups.GeneCount.tsv:每个物种中每个基因家族的数量。Orthogroup CG14 IR64 ZH11 Total OG0000000 0 298 4 302 OG0000001 0 3 217 220 OG0000002 0 0 107 107 OG0000003 0 2 85 87 OG0000004 0 83 0 83 OG0000005 2 30 43 75 OG0000006 53 3 3 59 OG0000007 0 33 26 59 OG0000008 1 55 1 57Orthogroups_SingleCopyOrthologues.txt:单拷贝基因家族编号。
单拷贝基因序列
Single_Copy_Orthologue_Sequences这个文件夹里面的是单拷贝基因家族的序列文件。每个文件就是一个单拷贝基因家族的序列。作用:单拷贝基因用于推断物种系统发育树。
Species_Tree
这个文件夹存放的是有根的物种树。
构建物种系统发育树
之前我使用的是OrthoFinder输出的物种树木,这里提供一种新的方法。
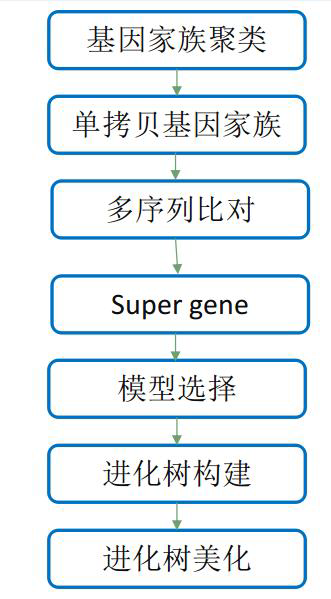
多序列比对
使用的是单拷贝基因。
ls 05.orthofinder/Single_Copy_Orthologue_Sequences/OG*[0-9].fa | awk '{print "mafft --localpair --maxiterate 1000 "$1" 1>"$1".mafft.fa 2>mafft.error"}' | sh
mkdir 06.species.tree
mv 05.orthofinder/Single_Copy_Orthologue_Sequences/*.mafft.fa ./06.species.tree/合并CDS序列
先合并为一个,再按照单拷贝基因家族进行拆分。点击下载脚本。
cat 03.cds.pep/*/Longest/*.longest.cds.fa > 06.species.tree/all.cds.fa
perl 01.scripts/get_single_copy_cds_file.pl 06.species.tree/all.cds.fa 05.orthofinder/Single_Copy_Orthologue_Sequences
mv 05.orthofinder/Single_Copy_Orthologue_Sequences/*.mafft.fa.cds ./06.species.tree
rm 06.species.tree/all.cds.fa 蛋白序列→密码子序列
ls 06.species.tree/OG*[0-9].fa.mafft.fa | awk '{print "pal2nal.pl "$1" "$1".cds -output fasta > "$1".codon.fa" }' | sh运行的过程中出现一些提示:
ERROR: number of input seqs differ (aa: 0; nuc: 3)!!
aa ''
nuc 'OsCG14G0780001574.01 OsIR64G0714247300.01 OsZH11G0717076700.01'
ERROR: number of input seqs differ (aa: 0; nuc: 3)!!
aa ''
nuc 'OsCG14G0713270300.01 OsIR64G0714248700.01 OsZH11G0717078300.01'比对结果优化
ls 06.species.tree/OG*[0-9].fa.mafft.fa.codon.fa | awk '{print "Gblocks "$1" -t=c -b5=h"}' | sh构建Super gene
需要注意物种编号:
cat 05.orthofinder/WorkingDirectory/SpeciesIDs.txt 0: CG14.fa
1: IR64.fa
2: ZH11.fa新建一个文件name.txt,顺序必须和上面的一致:
开始构建super gene(点击下载脚本):
perl 01.scripts/get_super_gene.pl 06.species.tree 06.species.tree/super.gene.phy.fa 06.species.tree/name.txt 
IQ-TREE建树
nohup iqtree -s 06.species.tree/super.gene.phy.fa -bb 1000 -m TEST -nt 1 -pre ./06.species.tree/IQ_TREE &出现报错,因为少于四个。
All model information printed to ./06.species.tree/IQ_TREE.model.gz
CPU time for ModelFinder: 4.087 seconds (0h:0m:4s)
Wall-clock time for ModelFinder: 4.695 seconds (0h:0m:4s)
Generating 1000 samples for ultrafast bootstrap (seed: 351912)...
ERROR: It makes no sense to perform bootstrap with less than 4 sequences.-rw-rw-r-- 1 lixiang lixiang 85 8月 9 10:49 06.species.tree/IQ_TREE.bionj
-rw-rw-r-- 1 lixiang lixiang 9.8K 8月 9 10:49 06.species.tree/IQ_TREE.ckp.gz
-rw-rw-r-- 1 lixiang lixiang 93 8月 9 10:49 06.species.tree/IQ_TREE.contree
-rw-rw-r-- 1 lixiang lixiang 8.0K 8月 9 10:49 06.species.tree/IQ_TREE.iqtree
-rw-rw-r-- 1 lixiang lixiang 19K 8月 9 10:49 06.species.tree/IQ_TREE.log
-rw-rw-r-- 1 lixiang lixiang 258 8月 9 10:49 06.species.tree/IQ_TREE.mldist
-rw-rw-r-- 1 lixiang lixiang 980 8月 9 10:49 06.species.tree/IQ_TREE.model.gz
-rw-rw-r-- 1 lixiang lixiang 280 8月 9 10:49 06.species.tree/IQ_TREE.splits.nex
-rw-rw-r-- 1 lixiang lixiang 93 8月 9 10:49 06.species.tree/IQ_TREE.treefile利用FigTree将Os设置为树的根。
全基因组比对和可视化
- 使用
minimap2进行基因组比对:
minimap2 -ax asm5 -t 75 --eqx nip.fa R498.chr12.fa | samtools sort -O BAM - > r498.2.nip.bam
minimap2 -ax asm5 -t 75 --eqx R498.chr12.fa acuce.chr12.fa | samtools sort -O BAM - > acuce2r498.bam 水稻基因组4分钟就能比对完。
- 使用
syri鉴定结构变异:
syri -c r498.2.nip.bam -r nip.fa -q R498.chr12.fa -F B --prefix r498.2.nip &
syri -c acuce2r498.bam -r R498.chr12.fa -q acuce.chr12.fa -F B --prefix acuce2r498 & - 准备
genomes.txt文件:
#file name
nip.fa Nipponbare
R498.chr12.fa R498
acuce.chr12.fa Acuce- 运行
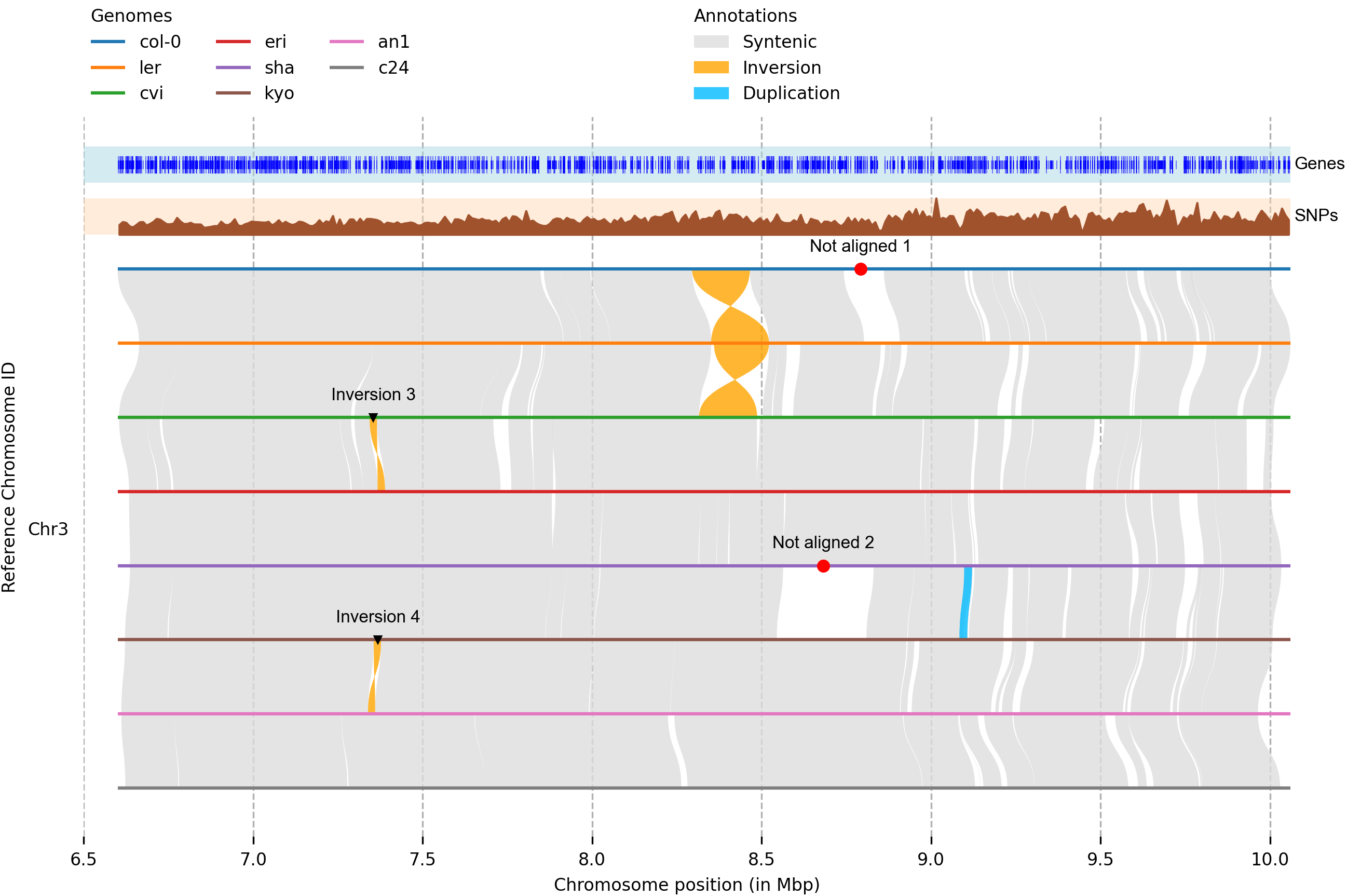
plotsr绘图:
plotsr --sr acuce2r498syri.out --genomes genomes.txt -o acuce2r498.plotsr.png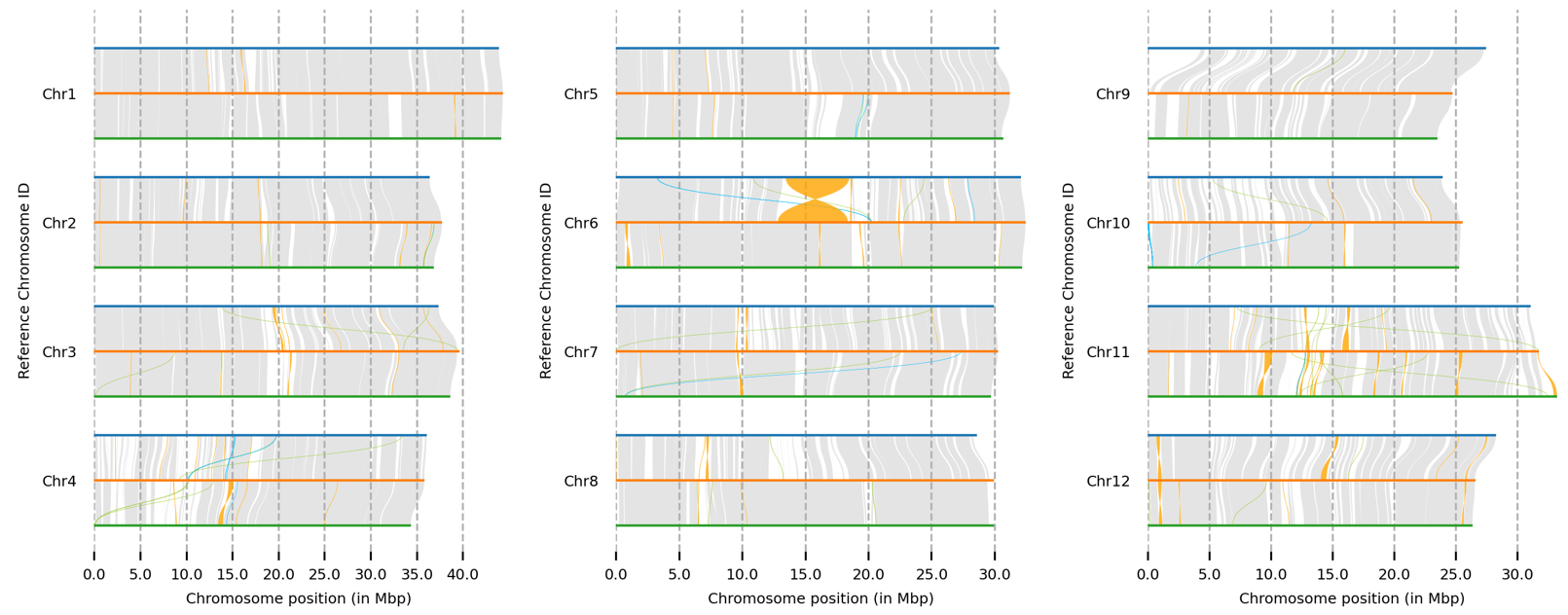
选择展示特定染色体:
plotsr --chr Chr5 --chr Chr6 --chr Chr7 --chr Chr8 --sr r498.2.nipsyri.out --sr acuce2r498syri.out --genomes genomes.txt -o nipponbare.r498.acuce.sr.plot.chr5-8.png群体遗传学
参考这个文章进行分析:
软件安装
基本上用mamba就能完成所有安装,比较坑的是GATK依赖的版本和其他软件的依赖有冲突,因此GATK需要单独创建一个环境。流程代码也需要修改,按大步骤分开运行。
基因组比对
主要分为两步:构建基因组索引和将测序数据比对到参考基因组上。
构建基因组索引
# bwa
bwa index 03.genome/genome.fa
# samtools
samtools faidx 03.genome/genome.fa
# picard
# picard CreateSequenceDictionary -R genome.fa
# GATK
gatk CreateSequenceDictionary -R 03.genome/genome.fa运行完成后得到的文件:
-rw-rw-r-- 1 lixiang lixiang 1.5K 8月 27 10:30 genome.dict
-rw-rw-r-- 1 lixiang lixiang 374M 8月 27 09:55 genome.fa
-rw-rw-r-- 1 lixiang lixiang 15 8月 27 10:48 genome.fa.amb
-rw-rw-r-- 1 lixiang lixiang 429 8月 27 10:48 genome.fa.ann
-rw-rw-r-- 1 lixiang lixiang 368M 8月 27 10:48 genome.fa.bwt
-rw-rw-r-- 1 lixiang lixiang 92M 8月 27 10:48 genome.fa.pac
-rw-rw-r-- 1 lixiang lixiang 184M 8月 27 10:50 genome.fa.sa
-rw-rw-r-- 1 lixiang lixiang 82M 8月 27 09:55 genome.gff3
-rw-rw-r-- 1 lixiang lixiang 71M 8月 27 10:00 genome.gtf
-rw-rw-r-- 1 lixiang lixiang 353 8月 27 10:25 samtools.fa.fai比对
将比对的结果直接传给samtools完成bam文件的排序:
bwa mem -M -t 50 -R "@RG\tID:s-100_H7GYJCCXY_L4\tSM:s-100_H7GYJCCXY_L4\tPL:illumina" 03.genome/genome.fa 02.data/yyt.reseq/s-100_H7GYJCCXY_L4_1.clean.fq.gz 02.data/yyt.reseq/s-100_H7GYJCCXY_L4_2.clean.fq.gz | samtools sort -@ 50 -m 8G -o 04.bwa.mapping/s-100_H7GYJCCXY_L4.sorted.bam-R这个参数必须有,不然后面GATK会报错。
耗时20分钟左右。
变异检测
主要是使用GATK4完成分析的。关于GATK4内存和线程数的使用,可以参考微信公众号文章。
标记重复
使用MarkDuplicates对重复序列进行标记。这个模块在GATK4和picard中都有。picard可以一步同时完成重复序列标记和构建索引,GATK需要分为两步。
picard
picard -Xmx400g MarkDuplicates I=04.bwa.mapping/s-100_H7GYJCCXY_L4.sorted.bam O=05.deduplicates/s-100_H7GYJCCXY_L4.sorted.bam CREATE_INDEX=true REMOVE_DUPLICATES=trueu M= 05.deduplicates/s-100_H7GYJCCXY_L4.txtGATK4
gatk --java-options "-Xmx20G -XX:ParallelGCThreads=8" MarkDuplicates --REMOVE_DUPLICATES false -I 04.bwa.mapping/s-100_H7GYJCCXY_L4.sorted.bam -O 05.deduplicates/s-100_H7GYJCCXY_L4.gatk.bam -M 05.deduplicates/s-100_H7GYJCCXY_L4.gatk.metrc.csv耗时9分钟左右。
构建bam文件索引:
samtools index -@ 20 05.deduplicates/s-100_H7GYJCCXY_L4.gatk.bam 变异检测
使用HaplotypeCaller模块进行变异检测,生成gvcf文件。只支持单样本。
gatk --java-options "-Xmx20G -XX:ParallelGCThreads=8" HaplotypeCaller -R 03.genome/genome.fa -I 05.deduplicates/s-100_H7GYJCCXY_L4.gatk.bam -O 06.haplotypecaller/s-100_H7GYJCCXY_L4.gvcf.gz -ERC GVCF从二代数据鉴定R基因
基因组下载
下载参考基因组数据。
get https://www.mbkbase.org/R498/R498_Chr.soft.fasta.gz
get https://www.mbkbase.org/R498/R498_IGDBv3_coreset.tar.gz
get https://www.mbkbase.org/R498/R498_CoreSet.pro.tar.gz
get https://www.mbkbase.org/R498/R498_CoreSet.pro.tar.gzBlast比对
核酸序列建库疯狂报错:
BLAST Database error: No alias or index file found for protein database [./blastdb/piapik] in search path [/sas16t/lixiang/yyt.reseq/02.blast::]换蛋白序列建库比对:
makeblastdb -in piapik.fa -dbtype prot -out blastdb/piapik
blastp -query R498_CoreSet.pros.format.fasta -db blastdb/piapik -evalue 1e-5 -num_threads 60 -outfmt 6 -out blast.out.txt提取gff文件和基因序列
提取gff文件:
grep "OsR498G1119642600.01" R498_IGDBv3_coreset/R498_IGDBv3_coreset.gff > pia.gff
grep "OsR498G1120737800.01" R498_IGDBv3_coreset/R498_IGDBv3_coreset.gff > pik.gff提取基因序列:
type="gene"
sed 's/"/\t/g' piapik.gff | awk -v type="${type}" 'BEGIN{OFS=FS="\t"}{if($3==type) {print $1,$4-1,$5,$14,".",$7}}' > piapik.bed 提取蛋白序列和CDS序列用于后续的SnpEff:
seqkit grep -f piapik.cds.id.txt genome.cds.fa > piapik.cds.fa
seqkit grep -f piapik.cds.id.txt genome.pro.fa > piapik.pro.fa将这些文件拷贝到SnpEff文件夹:
cp ../../../01.data/genome/piapik.fa sequences.fa
cp ../../../01.data/genome/piapik.gff genes.gff
cp ../../../01.data/genome/R498_IGDBv3_coreset/piapik.cds.fa cds.fa
cp ../../../01.data/genome/R498_IGDBv3_coreset/piapik.pro.fa ./protein.fa
cp PiaPik/sequences.fa ./genomes/PiaPik.fa 构建索引
bwa index 01.data/genome/piapik.fa
samtools faidx 01.data/genome/piapik.fa
gatk CreateSequenceDictionary -R 01.data/genome/piapik.fa SnpEff索引
java -jar /home/lixiang/mambaforge/envs/gatk4/share/snpeff-5.1-2/snpEff.jar build -c snpEff.config -gff3 -v PiaPik -noCheckCds -noCheckProtein批量运行
rm(list = ls())
library(tidyverse)
dir("./01.data/clean.data/") %>%
as_data_frame() %>%
magrittr::set_names("file") %>%
dplyr::mutate(sample = stringr::str_split(file, "\\.") %>%
sapply("[", 1) %>%
stringr::str_sub(1, stringr::str_length(.)-2)) -> samples
### 构建批量代码
genome = "01.data/genome/piapik.fa "
all.run = NULL
for (i in unique(samples$sample)) {
### exit then skip
if (file.exists(sprintf("./03.mapping/%s.sorted.mapped.bam",i))) {
next
}else{
f.read = sprintf("./01.data/clean.data/%s_1.clean.fq.gz",i)
r.read = sprintf("./01.data/clean.data/%s_2.clean.fq.gz",i)
title = sprintf("\"@RG\\tID:%s\\tSM:%s\\tPL:illumina\"",i,i)
bam = sprintf("./03.mapping/%s.sorted.bam",i)
### bwa mem
sprintf("bwa mem -M -t 75 -R %s %s %s %s | samtools sort -@ 50 -m 8G -o %s",title, genome, f.read, r.read, bam) %>%
as_data_frame() %>%
rbind(all.run) -> all.run
### extract mapped
sprintf("samtools view -bF 12 -@ 75 03.mapping/%s.sorted.bam -o 04.sorted.bam/%s.sorted.mapped.bam",i,i) %>%
as_data_frame() %>%
rbind(all.run) -> all.run
### index
sprintf("samtools index -@ 75 -bc 04.sorted.bam/%s.sorted.mapped.bam",i) %>%
as_data_frame() %>%
rbind(all.run) -> all.run
### ### Pia
### sprintf("samtools view 03.mapping/%s.sorted.mapped.bam Pia -o 03.mapping/%s.sorted.mapped.Pia.bam",i,i) %>%
### as_data_frame() %>%
### rbind(all.run) -> all.run
###
### ### Pik
### sprintf("samtools view 03.mapping/%s.sorted.mapped.bam Pik -o 03.mapping/%s.sorted.mapped.Pik.bam",i,i) %>%
### as_data_frame() %>%
### rbind(all.run) -> all.run
### ### bam2fastq
### sprintf("bedtools bamtofastq -i 03.mapping/%s.sorted.mapped.Pia.bam -fq 03.mapped.fastq/%s_1.Pia.fsatq -fq2 03.mapped.fastq/%s_2.Pia.fsatq",i,i,i) %>%
### as_data_frame() %>%
### rbind(all.run) -> all.run
### sprintf("bedtools bamtofastq -i 03.mapping/%s.sorted.mapped.Pik.bam -fq 03.mapped.fastq/%s_1.Pik.fsatq -fq2 03.mapped.fastq/%s_2.Pik.fsatq",i,i,i) %>%
### as_data_frame() %>%
### rbind(all.run) -> all.run
### ### rm bam
### sprintf("rm 03.mapping/%s.sorted.bam",i) %>%
### as_data_frame() %>%
### rbind(all.run) -> all.run
### coverage
sprintf("samtools coverage ./04.sorted.bam/%s.sorted.mapped.bam > 05.converage/%s.coverage.txt",i,i) %>%
as_data_frame() %>%
rbind(all.run) -> all.run
### picard
sprintf("picard -Xmx400g MarkDuplicates I=04.sorted.bam/%s.sorted.mapped.bam O=06.picard/%s.picard.bam CREATE_INDEX=true REMOVE_DUPLICATES=trueu M=06.picard/%s.txt",i, i, i) %>%
as_data_frame() %>%
rbind(all.run) -> all.run
### gatk
sprintf("gatk --java-options \"-Xmx20G -XX:ParallelGCThreads=8\" HaplotypeCaller -R 01.data/genome/piapik.fa -I 06.picard/%s.picard.bam -O 07.gatk/%s.gvcf -ERC GVCF",i,i) %>%
as_data_frame() %>%
rbind(all.run) ->all.run
### gvcf 2 vcf
sprintf("gatk GenotypeGVCFs -R 01.data/genome/piapik.fa -V 07.gatk/%s.gvcf -O 07.gatk/%s.vcf",i,i) %>%
as_data_frame() %>%
rbind(all.run) ->all.run
### SneEff
sprintf("java -Xmx10G -jar ~/mambaforge/envs/gatk4/share/snpeff-5.1-2/snpEff.jar ann -c 08.snpeff/snpEff.config PiaPik 07.gatk/%s.vcf > 08.snpeff/%s.ann.vcf -csvStats 08.snpeff/%s.positive.csv -stats 08.snpeff/%s.positive.html",i,i,i,i) %>%
as_data_frame() %>%
rbind(all.run) ->all.run
}
}
all.run %>%
dplyr::slice(nrow(all.run):1) %>%
write.table("./run.all.sh", col.names = FALSE, row.names = FALSE, quote = FALSE)合并覆盖度
dir("./05.converage/") %>%
as_data_frame() %>%
magrittr::set_names("file") -> coverage
all.converage.final = NULL
for (i in coverage$file) {
sprintf("./05.converage/%s",i) %>%
readr::read_delim() %>%
dplyr::mutate(sample = stringr::str_replace(i,".coverage.txt","")) %>%
dplyr::select(ncol(.), 1:(ncol(.)-1)) %>%
rbind(all.converage.final) -> all.converage.final
}
all.converage.final %>%
write.table("./all.coverage.txt", sep = "\t", quote = FALSE, row.names = FALSE)判断突变类型
rm(list = ls())
readr::read_delim("./01.data/genome/piapik.gff", col_names = FALSE) %>%
dplyr::select(1,3:5) %>%
magrittr::set_names(c("HROM","gene.region","start","end")) %>%
dplyr::filter(gene.region == "exon") %>%
dplyr::mutate(HROM = stringr::str_replace(HROM,"\\.","_"))-> df.gff
dir("./08.snpeff/") %>%
as_data_frame() %>%
magrittr::set_names("vcf") %>%
dplyr::filter(stringr::str_ends(vcf, "ann.vcf"))-> vcf
all.vcf = NULL
for (i in vcf$vcf) {
sprintf("./08.snpeff/%s",i) %>%
data.table::fread() %>%
magrittr::set_names(c("HROM", "POS", "ID", "REF", "ALT", "QUAL", "FILTER", "INFO", "FORMAT", "sample")) %>%
dplyr::select(1:8) %>%
dplyr::mutate(mutation.type = stringr::str_split(INFO,";") %>%
sapply("[",12) %>%
stringr::str_split("\\|") %>%
sapply("[",2),
mutation.region = stringr::str_split(INFO,";") %>%
sapply("[",12) %>%
stringr::str_split("\\|") %>%
sapply("[",6),
mutation.protein = stringr::str_split(INFO,";") %>%
sapply("[",12) %>%
stringr::str_split("\\|") %>%
sapply("[",11)) %>%
dplyr::select(-INFO) %>%
dplyr::left_join(df.gff) %>%
dplyr::mutate(group = case_when(POS >= start & POS <= end ~ "Exon", TRUE ~ "Non-exon")) %>%
dplyr::filter(group == "Exon") %>%
dplyr::select(1:5,8:10) %>%
magrittr::set_names(c("chrom", "position", "id", "ref", "alt", "mutation.type", "mutation.region", "mutation.protein")) %>%
dplyr::mutate(sample = stringr::str_replace(i,".ann.vcf","")) %>%
dplyr::mutate(rgene = case_when(chrom == "OsR498G1119642600_01" ~ "Pia", TRUE ~ "Pik")) %>%
dplyr::select(9:10,2:8) %>%
rbind(all.vcf) -> all.vcf
}
all.vcf %>%
write.table("./all.vcf.txt", sep = "\t", quote = FALSE, row.names = FALSE)扩增子
软件安装
数据处理
数据抽平
# df.kraken2.raw.count的行是特征变量,列是样品
vegan::rrarefy(
t(df.kraken2.raw.count),
min(colSums(df.kraken2.raw.count))
) %>%
t() %>%
as.data.frame() -> df宏基因组
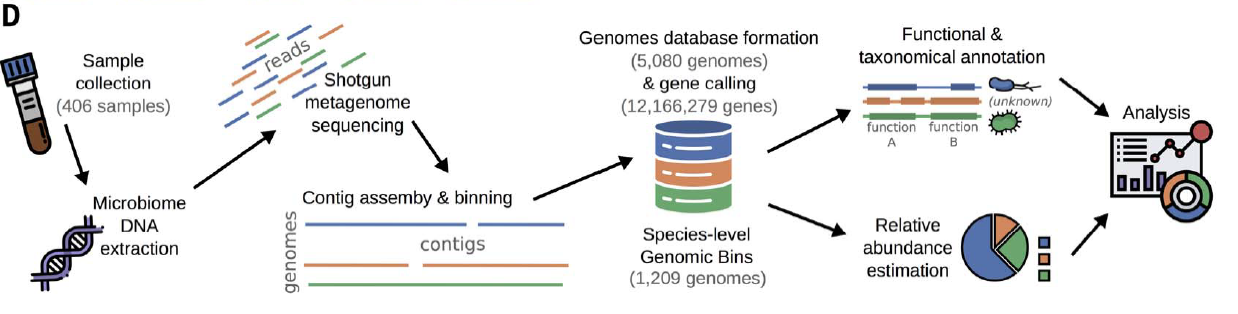
软件安装
mamba install -c bioconda kraken2
mamba install -c bioconda checkm2
mamba install -c bioconda drep
mamba install -c bioconda gtdbtk=2.1.1
mamba install -c bioconda quast数据库配置
Kraken2数据库
可以直接从国家微生物数据科学中心下载,下载链接:ftp://download.nmdc.cn/tools/meta/kraken2/. 下载完成直接解压可以使用了:
rw-r--r-- 1 lixiang lixiang 3.4M 3月 11 04:37 database100mers.kmer_distrib
-rw-r--r-- 1 lixiang lixiang 3.1M 3月 11 08:58 database150mers.kmer_distrib
-rw-r--r-- 1 lixiang lixiang 2.8M 3月 11 14:27 database200mers.kmer_distrib
-rw-r--r-- 1 lixiang lixiang 2.6M 3月 11 20:26 database250mers.kmer_distrib
-rw-r--r-- 1 lixiang lixiang 2.4M 3月 12 03:13 database300mers.kmer_distrib
-rw-r--r-- 1 lixiang lixiang 3.9M 3月 10 22:23 database50mers.kmer_distrib
-rw-r--r-- 1 lixiang lixiang 3.6M 3月 11 01:10 database75mers.kmer_distrib
-rw-r--r-- 1 lixiang lixiang 69G 3月 10 19:33 hash.k2d
-rw-r--r-- 1 lixiang lixiang 2.7M 3月 10 19:41 inspect.txt
-rw-rw-r-- 1 lixiang lixiang 53G 6月 14 13:01 k2_pluspf_20230314.tar.gz
-rw-r--r-- 1 lixiang lixiang 2.1M 3月 10 19:42 ktaxonomy.tsv
-rw-r--r-- 1 lixiang lixiang 64 3月 10 19:33 opts.k2d
-rw-r--r-- 1 lixiang lixiang 6.0M 3月 10 09:12 seqid2taxid.map
-rw-r--r-- 1 lixiang lixiang 3.2M 3月 10 09:42 taxo.k2d
drwxrwxr-x 3 lixiang lixiang 4.0K 6月 15 18:18 taxonomyCheckM2数据库
参考文献:
公众号解读:
根据官方文档直接使用下面的命令下载配置数据库:
checkm2 database --download --path ~/database/checkm2下载速度还行,校园网可以达到5M/s左右。
下载完成后使用下面的代码测试有没有安装配置成功:
checkm2 testrun没问题的话会输出:
[08/13/2023 09:35:33 AM] INFO: Test run: Running quality prediction workflow on test genomes with 50 threads.
[08/13/2023 09:35:33 AM] INFO: Running checksum on test genomes.
[08/13/2023 09:35:33 AM] INFO: Checksum successful.
[08/13/2023 09:35:36 AM] INFO: Calling genes in 3 bins with 50 threads:
Finished processing 3 of 3 (100.00%) bins.
[08/13/2023 09:36:01 AM] INFO: Calculating metadata for 3 bins with 50 threads:
Finished processing 3 of 3 (100.00%) bin metadata.
[08/13/2023 09:36:03 AM] INFO: Annotating input genomes with DIAMOND using 50 threads
[08/13/2023 09:36:55 AM] INFO: Processing DIAMOND output
[08/13/2023 09:36:55 AM] INFO: Predicting completeness and contamination using ML models.
[08/13/2023 09:37:05 AM] INFO: Parsing all results and constructing final output table.
[08/13/2023 09:37:05 AM] INFO: CheckM2 finished successfully.
[08/13/2023 09:37:05 AM] INFO: Test run successful! See README for details. Results:
Name Completeness Contamination Completeness_Model_Used Translation_Table_Used
TEST1 100.00 0.74 Neural Network (Specific Model) 11
TEST2 98.54 0.21 Neural Network (Specific Model) 11
TEST3 98.75 0.51 Neural Network (Specific Model) 11手动配置现有数据库:
checkm data setRoot /sas16t/lixiang/database/checkmGTDB-TK数据库
安装完成后会提示配置数据库:
Automatic:
1. Run the command "download-db.sh" to automatically download and extract to:
/home/user/mambaforge/envs/metagenome/share/gtdbtk-2.1.1/db/
Manual:
1. Manually download the latest reference data:
wget https://data.gtdb.ecogenomic.org/releases/release207/207.0/auxillary_files/gtdbtk_r207_v2_data.tar.gz
2. Extract the archive to a target directory:
tar -xvzf gtdbtk_r207_v2_data.tar.gz -C "/path/to/target/db" --strip 1 > /dev/null
rm gtdbtk_r207_v2_data.tar.gz
3. Set the GTDBTK_DATA_PATH environment variable by running:
conda env config vars set GTDBTK_DATA_PATH="/path/to/target/db"
由于mamba是安装在home目录下的,数据库比较占空间,所以手动下载数据库放到其他盘,然后在.zshrc文件中写这样一句配置(PS:尝试过将数据库软连接到/home/lixiang/mambaforge/envs/metagenome/share/gtdbtk-2.1.1/db/,没有成功):
GTDBTK_DATA_PATH="/sas16t/user/database/gtdb.v207/"然后进行测试是否配置成功:
>gtdbtk test
[2023-08-13 16:26:22] INFO: GTDB-Tk v2.3.0
[2023-08-13 16:26:22] INFO: gtdbtk test
[2023-08-13 16:26:23] INFO: Using GTDB-Tk reference data version r214: /sas16t/lixiang/database/gtdb.v207/
[2023-08-13 16:26:23] INFO: Using a temporary directory as out_dir was not specified.
[2023-08-13 16:26:23] INFO: Command: gtdbtk classify_wf --skip_ani_screen --genome_dir /tmp/gtdbtk_tmp_wc9jpdog/genomes --out_dir /tmp/gtdbtk_tmp_wc9jpdog/output --cpus 1 -f
<TEST OUTPUT> [2023-08-13 16:26:23] INFO: gtdbtk classify_wf --skip_ani_screen --genome_dir /tmp/gtdbtk_tmp_wc9jpdog/genomes --out_dir /tmp/gtdbtk_tm <TEST OUTPUT> [2023-08-13 16:26:23] INFO: Using GTDB-Tk reference data version r214: /sas16t/lixiang/database/gtdb.v207/ <TEST OUTPUT> [2023-08-13 16:40:26] INFO: Note that Tk classification mode is insufficient for publication of new taxonomic designations. New designa <TEST OUTPUT> [2023-08-13 16:40:26] INFO: Done.
[2023-08-13 16:40:26] INFO: Test has successfully finished. 如果配置失败的话可以使用下面的代码进行配置:
mamba env config vars set GTDBTK_DATA_PATH="/sas16t/lixiang/database/gtdb.v207/"再运行gtdbtk check_install验证是否安装成功:
[2023-09-07 15:19:51] INFO: GTDB-Tk v2.3.0
[2023-09-07 15:19:51] INFO: gtdbtk check_install
[2023-09-07 15:19:51] INFO: Using GTDB-Tk reference data version r214: /sas16t/lixiang/database/gtdb.v207/
[2023-09-07 15:19:51] INFO: Running install verification
[2023-09-07 15:19:51] INFO: Checking that all third-party software are on the system path:
[2023-09-07 15:19:51] INFO: |-- FastTree OK
[2023-09-07 15:19:51] INFO: |-- FastTreeMP OK
[2023-09-07 15:19:51] INFO: |-- fastANI OK
[2023-09-07 15:19:51] INFO: |-- guppy OK
[2023-09-07 15:19:51] INFO: |-- hmmalign OK
[2023-09-07 15:19:51] INFO: |-- hmmsearch OK
[2023-09-07 15:19:51] INFO: |-- mash OK
[2023-09-07 15:19:51] INFO: |-- pplacer OK
[2023-09-07 15:19:51] INFO: |-- prodigal OK
[2023-09-07 15:19:51] INFO: Checking integrity of reference package: /sas16t/lixiang/database/gtdb.v207/
[2023-09-07 15:19:54] INFO: |-- pplacer OK
[2023-09-07 15:19:54] INFO: |-- masks OK
[2023-09-07 15:19:54] INFO: |-- markers OK
[2023-09-07 15:19:55] INFO: |-- radii OK
[2023-09-07 15:20:09] INFO: |-- msa OK
[2023-09-07 15:20:09] INFO: |-- metadata OK
[2023-09-07 15:20:09] INFO: |-- taxonomy OK
[2023-09-07 15:27:54] INFO: |-- fastani OK
[2023-09-07 15:27:54] INFO: |-- mrca_red OK
[2023-09-07 15:27:54] INFO: Done.还需要配置Mash数据库,数据库下载地址:https://figshare.com/articles/online_resource/Mash_Sketched_databases_for_Accessible_Reference_Data_for_Genome-Based_Taxonomy_and_Comparative_Genomics/14408801
下载解压后放在GTDB数据库下的mash文件夹内即可。
-rw-rw-r-- 1 lixiang lixiang 138M 9月 7 15:38 Bacteria_Archaea_type_assembly_set.msh
-rw-rw-r-- 1 lixiang lixiang 3.2M 9月 7 15:38 Bacteria_Archaea_type_proteome_set.msh
-rw-rw-r-- 1 lixiang lixiang 4.9M 9月 7 15:38 Freshwater_Metagenome_assembly_set.msh
-rw-rw-r-- 1 lixiang lixiang 78M 9月 7 15:38 Fungal_Database.2022_genomic.fna.gz.msh
-rw-rw-r-- 1 lixiang lixiang 6.4M 9月 7 15:38 Fungi_type_assembly_set.msh
-rw-rw-r-- 1 lixiang lixiang 88K 9月 7 15:38 Fungi_type_proteome_set.msh
-rw-rw-r-- 1 lixiang lixiang 377M 9月 7 15:38 GTDBr202_genomic.fna.msh
-rw-rw-r-- 1 lixiang lixiang 3.8M 9月 7 15:38 Soil_Metgenome_assembly_set.msh
-rw-rw-r-- 1 lixiang lixiang 352M 9月 7 15:38 Virus_Sept21_GenBank_assembly_set.mshKraken
数据库配置查看上一节。
使用方法如下:
kraken2 --db /path/to/kraken2数据库 --threads 50 --report sample.kraken2.report --report-minimizer-data --minimum-hit-groups 2 sample.r1.fq.gz sample.r2.fq.gz > sample.kraken2从report中提出结果:
awk '$6 == "S" {print}' sample.kraken2.report > sample.kraken2.count.txt 0.05 36989 36989 145318 94971 S 2782665 Bradyrhizobium sp. 200
0.05 35410 35410 136532 88732 S 2782641 Bradyrhizobium sp. 170
0.03 26027 26027 120680 75200 S 1325120 Bradyrhizobium sp. CCBAU 53421
0.03 22196 22196 80115 52688 S 858422 Bradyrhizobium sp. CCBAU 051011
0.03 20407 20407 75697 50707 S 1325100 Bradyrhizobium sp. CCBAU 51753
0.03 19362 19362 117767 78227 S 1197460 Bradyrhizobium sp. 6(2017)
0.02 17401 17401 66466 42997 S 2057741 Bradyrhizobium sp. SK17
0.02 15609 15609 72888 45484 S 1404443 Bradyrhizobium sp. 41S5
0.01 11111 11111 36661 22319 S 1521768 Bradyrhizobium sp. WD16
0.01 10672 10672 54472 41285 S 1325114 Bradyrhizobium sp. CCBAU 53351包括 6 列,方便整理下游分析。
百分比
count
count 最优
(U) nclassified, (R) oot, (D) omain, (K) ingdom, (P) hylum, (C) lass, (O) rder, (F) amily, (G) enus, or (S) pecies. “G2” 代表位于属一种间
NCBI 物种 ID
科学物种名
加了参数--report-minimizer-data后会多输出两列,也就是第四列和第五列。
也可以搭配bracken使用:
bracken -d /path/to/kraken2数据库 -l S -r 100 -t 50 -i sample.kraken2.report -o sample.bracken -w sample.brackenw输出结果:
name taxonomy_id taxonomy_lvl kraken_assigned_reads added_reads new_est_reads fraction_total_reads
Bradyrhizobium sp. 200 2782665 S 36989 8041 45030 0.00301
Bradyrhizobium sp. 170 2782641 S 35410 9381 44791 0.00299
Bradyrhizobium sp. CCBAU 53421 1325120 S 26027 6399 32426 0.00216
Bradyrhizobium sp. CCBAU 051011 858422 S 22196 3166 25362 0.00169
Bradyrhizobium sp. CCBAU 51753 1325100 S 20407 3304 23711 0.00158
Bradyrhizobium sp. 6(2017) 1197460 S 19362 7354 26716 0.00178
Bradyrhizobium sp. SK17 2057741 S 17401 6079 23480 0.00157
Bradyrhizobium sp. 41S5 1404443 S 15609 8526 24135 0.00161
Bradyrhizobium sp. WD16 1521768 S 11111 1356 12467 0.00083组装和评估
组装直接使用MetaWRAP的功能模块即可:
metawrap assembly -1 ./03.each.sample/SRR9948796/2.clean.reads/SRR9948796_1.fastq -2 ./03.each.sample/SRR9948796/2.clean.reads/SRR9948796_2.fastq -m 200 -t 60 --megahit -o ./03.each.sample/SRR9948796/3.assembly质量评估使用QUAST:
quast -t 70 ./03.each.sample/SRR9948809/3.assembly/final_assembly.fasta -o ./09.assembly.quality/SRR9948809基因丰度
使用prodigal完成基因预测:
prodigal -p meta -a ./10.assembly.gene.prediction/SRR16798125.final_assembly.pep -d ./10.assembly.gene.prediction/SRR16798125.final_assembly.cds -f gff -g 11 -o ./10.assembly.gene.prediction/SRR16798125.final_assembly.gff -i ./03.each.sample/SRR16798125/3.assembly/final_assembly.fasta > ./10.assembly.gene.prediction/SRR16798125.prodigal.log分箱策略
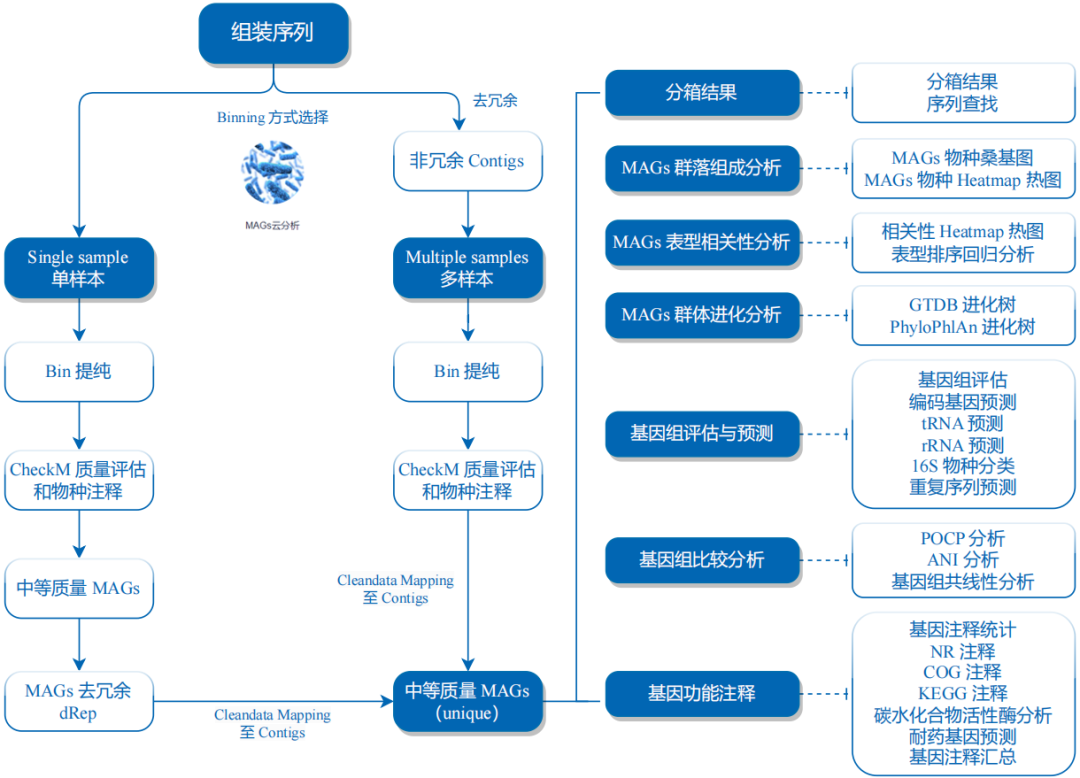
总体思路:三种不同的分箱软件→分箱优化→去重→分类注释和功能注释。
分箱质检
CheckM2
checkm2 predict --threads 50 -x .fa --input 07.all.init.bins/bins/ --output-directory 07.all.init.bins/checkm2 > checkm2.log 其中:
-x表示后缀,默认的是.fna.
分箱聚类
fastANI
平均核苷酸一致性Average Nucleotide Identity (ANI),参考下面这篇文献使用fastANI:
fastANI -t 70 --rl 15.bins.ANI/all.bins.id.txt --ql 15.bins.ANI/all.bins.id.txt -o 15.bins.ANI/all.bins.fastANI.txt2400对,跑了36分钟。
用ANI聚类的阈值是89:
The cutoff for hierarchical clustering was set while minimizing both over
and under clustering for ANI of 89.
ANI大于95的bins进行合并(但是如果分类注释的时候是不同的分类水平则拆分开):
Then, clusters for which all bins had ANI greater than 95 were merged.
每个cluster代表性的bins如何挑选呢:
For each cluster the representative genome was set based on the following
metrics: completeness, contamination, coverage, polymorphism and N50. Each genome in a cluster was scored based on the percentile of this genome across all the 1,582 genomes. The genome with the highest score was set as the representative genome.
在这个文献中还提到了根据ANI的值来判断MAG是全新的还是已经被报道过的。
The maximum ANI values were used for novelty categorization as follows: rSGBs with maximum ANI values above 95% were defined as known species, values below 83% as previously undescribed (novel) species, and values in between as intermediate.
可以以proGenomes(一共有4305248个contigs)作为参考基因组,参考文献:
使用案例:
fastANI -q ../13.bins.class/data/SRR10695388_concoct_bin.1.fa -r /sas16t/lixiang/database/progenomes/progenomes3.contigs.representatives.fasta -
o ani.res.txt分箱注释
GTDB
使用GTDB进行注释:
gtdbtk classify_wf --cpus 60 --mash_db /sas16t/lixiang/database/gtdb.v207/mash -x fa --genome_dir data --out_dir ./输出这些文件:
drwxrwxr-x 2 lixiang lixiang 4.0K 9月 7 15:59 align
drwxrwxr-x 2 lixiang lixiang 4.0K 9月 7 15:59 classify
drwxrwxr-x 2 lixiang lixiang 4.0K 9月 7 15:32 data
lrwxrwxrwx 1 lixiang lixiang 32 9月 7 15:44 gtdbtk.ar53.summary.tsv -> classify/gtdbtk.ar53.summary.tsv
lrwxrwxrwx 1 lixiang lixiang 34 9月 7 15:59 gtdbtk.bac120.summary.tsv -> classify/gtdbtk.bac120.summary.tsv
-rw-rw-r-- 1 lixiang lixiang 3.5K 9月 7 15:59 gtdbtk.json
-rw-rw-r-- 1 lixiang lixiang 6.6K 9月 7 15:59 gtdbtk.log
-rw-rw-r-- 1 lixiang lixiang 0 9月 7 15:36 gtdbtk.warnings.log
drwxrwxr-x 2 lixiang lixiang 4.0K 9月 7 15:59 identify构建系统发育树:
gtdbtk infer --cpus 60 --msa_file align/gtdbtk.bac120.user_msa.fasta.gz --out_dir ./drwxrwxr-x 2 lixiang lixiang 4.0K 9月 7 15:59 align
drwxrwxr-x 2 lixiang lixiang 4.0K 9月 7 15:59 classify
drwxrwxr-x 2 lixiang lixiang 4.0K 9月 7 15:32 data
lrwxrwxrwx 1 lixiang lixiang 32 9月 7 15:44 gtdbtk.ar53.summary.tsv -> classify/gtdbtk.ar53.summary.tsv
lrwxrwxrwx 1 lixiang lixiang 34 9月 7 15:59 gtdbtk.bac120.summary.tsv -> classify/gtdbtk.bac120.summary.tsv
-rw-rw-r-- 1 lixiang lixiang 718 9月 7 16:02 gtdbtk.json
-rw-rw-r-- 1 lixiang lixiang 7.1K 9月 7 16:02 gtdbtk.log
lrwxrwxrwx 1 lixiang lixiang 47 9月 7 16:02 gtdbtk.unrooted.tree -> infer/intermediate_results/gtdbtk.unrooted.tree
-rw-rw-r-- 1 lixiang lixiang 0 9月 7 15:36 gtdbtk.warnings.log
drwxrwxr-x 2 lixiang lixiang 4.0K 9月 7 15:59 identify
drwxrwxr-x 3 lixiang lixiang 4.0K 9月 7 16:02 infergtdbtk.unrooted.tree就是需要的树文件 。
分箱去重
有时候分箱得到的结果是非冗余的,也就是部分bins基本是一样的,这时候就需要去重。可以使用dRep进行去重。dRep参考文献:
有几个软件比较难安装:
ANIcalculator:https://ani.jgi.doe.gov/html/anicalculator.php. 下载了解压就行。nsimscan:https://github.com/abadona/qsimscan. 这个需要编译。Centrifuge:https://github.com/DaehwanKimLab/centrifuge. 这个也需要编译:git clone https://github.com/DaehwanKimLab/centrifuge cd centrifuge make
使用方法:
dRep dereplicate drep.res -g data/*.fa输出的提示信息:
Dereplicated genomes................. /home/lixiang/project/pn.metagenomics/temp/drep.res/dereplicated_genomes/
Dereplicated genomes information..... /home/lixiang/project/pn.metagenomics/temp/drep.res/data_tables/Widb.csv
Figures.............................. /home/lixiang/project/pn.metagenomics/temp/drep.res/figures/
Warnings............................. /home/lixiang/project/pn.metagenomics/temp/drep.res/log/warnings.txt目录dereplicated_genomes是去重后的bins.
分箱建树
使用PhyloPhlAn 3.0构建系统发育树。参考文献:
下载数据库
PhyloPhlAn (
-d phylophlan, 400 universal marker genes) presented in Segata, N et al. NatComm 4:2304 (2013)AMPHORA2 (
-d amphora2, 136 universal marker genes) presented in Wu M, Scott AJ Bioinformatics 28.7 (2012)
下载了直接解压就行:
http://cmprod1.cibio.unitn.it/databases/PhyloPhlAn/amphora2.tar
http://cmprod1.cibio.unitn.it/databases/PhyloPhlAn/phylophlan.tar生成配置文件
通常是使用蛋白序列进行建树,类似于OrthoFinder。
phylophlan_write_config_file -o 17.bins.tree/config_aa.cfg --force_nucleotides -d a --db_aa diamond --map_aa diamond --msa mafft --trim trimal --tree1 fasttree --tree2 raxml --overwrite配置文件示例:
[db_aa]
program_name = /home/lixiang/mambaforge/envs/metagenome/bin/diamond
params = makedb
threads = --threads
input = --in
output = --db
version = version
command_line = #program_name# #params# #threads# #input# #output#
[map_aa]
program_name = /home/lixiang/mambaforge/envs/metagenome/bin/diamond
params = blastp --quiet --threads 60 --outfmt 6 --more-sensitive --id 50 --max-hsps 35 -k 0
input = --query
database = --db
output = --out
version = version
command_line = #program_name# #params# #input# #database# #output#
[msa]
program_name = /home/lixiang/mambaforge/envs/metagenome/bin/mafft
params = --quiet --anysymbol --thread 60 --auto
version = --version
command_line = #program_name# #params# #input# > #output#
environment = TMPDIR=/tmp
[trim]
program_name = /home/lixiang/mambaforge/envs/metagenome/bin/trimal
params = -gappyout
input = -in
output = -out
version = --version
command_line = #program_name# #params# #input# #output#
[tree1]
program_name = /home/lixiang/mambaforge/envs/metagenome/bin/FastTreeMP
params = -quiet -pseudo -spr 4 -mlacc 2 -slownni -fastest -no2nd -mlnni 4 -gtr -nt
output = -out
command_line = #program_name# #params# #output# #input#
environment = OMP_NUM_THREADS=3
[tree2]
program_name = /home/lixiang/mambaforge/envs/metagenome/bin/raxmlHPC-PTHREADS-SSE3
params = -p 1989 -m GTRCAT
database = -t
input = -s
output_path = -w
output = -n
version = -v
command_line = #program_name# #params# #threads# #database# #output_path# #input# #output#
threads = -T构建进化树
nohup phylophlan -d phylophlan --databases_folder ~/lixiang/database/phylophlan3/ -t a -f 17.bins.tree/config_aa.cfg --nproc 60 --diversity medium --fast -i 17.bins.tree/pep -o 17.bins.tree > phylophlan.log 2>&1 &系统发育树可视化使用iTOL完成。
MetaWRAP流程
参考文献
软件安装
软件地址:https://github.com/bxlab/metaWRAP. 软件的依赖非常非常多,我是这样安装成功的:
mamba create -y --name metawrap-env --channel ursky metawrap-mg=1.3.2检查是否安装成功:
> metawrap -h
MetaWRAP v=1.3.2
Usage: metaWRAP [module]
Modules:
read_qc Raw read QC module (read trimming and contamination removal)
assembly Assembly module (metagenomic assembly)
kraken KRAKEN module (taxonomy annotation of reads and assemblies)
kraken2 KRAKEN2 module (taxonomy annotation of reads and assemblies)
blobology Blobology module (GC vs Abund plots of contigs and bins)
binning Binning module (metabat, maxbin, or concoct)
bin_refinement Refinement of bins from binning module
reassemble_bins Reassemble bins using metagenomic reads
quant_bins Quantify the abundance of each bin across samples
classify_bins Assign taxonomy to genomic bins
annotate_bins Functional annotation of draft genomes
--help | -h show this help message
--version | -v show metaWRAP version
--show-config show where the metawrap configuration files are stored数据库配置
CheckM
mkcd ~/database/checkm
axel -n 50 ftp://download.nmdc.cn/tools/meta/checkm/checkm_data_2015_01_16.tar.gz
tar -xvf checkm_data_2015_01_16.tar.gz
# 输入下面代码配置数据库
checkm data setRootKraken2
mkcd ~/database/kraken2
axel -n 50 ftp://download.nmdc.cn/tools/meta/kraken2/k2_pluspf_20230314.tar.gz
tar -xvf k2_pluspf_20230314.tar.gzNCBI-nt
mkcd ~/database/ncbi.nt
~/.aspera/connect/bin/ascp -i ~/mambaforge/envs/tools4bioinf/etc/asperaweb_id_dsa.openssh --overwrite=diff -QTr -l6000m \
anonftp@ftp.ncbi.nlm.nih.gov:blast/db/v4/nt_v4.{00..85}.tar.gz ./NCBI-taxonomy
mkcd ~/database/ncbi.taxonomy
axel -n 50 ftp://ftp.ncbi.nlm.nih.gov/pub/taxonomy/taxdump.tar.gz
tar -xvf taxdump.tar.gz宿主基因组
以人类的hg38基因组为例。
mkcd ~/database/human.genome
axel -n 50 http://hgdownload.soe.ucsc.edu/goldenPath/hg38/bigZips/hg38.fa.gz
gunzip hg38.fa.gz
cat *fa > hg38.fa
rm chr*.fa
bmtool -d hg38.fa -o hg38.bitmask
srprism mkindex -i hg38.fa -o hg38.srprism -M 100000修改配置文件
上述数据库下载完成后需要修改MetaWRAP的配置文件,不需要的就注释掉,路径一定要写对,不然会报错的。
vi ~/mambaforge/envs/metawrap-env/bin/config-metawrap
# Paths to metaWRAP scripts (dont have to modify)
mw_path=$(which metawrap)
bin_path=${mw_path%/*}
SOFT=${bin_path}/metawrap-scripts
PIPES=${bin_path}/metawrap-modules
# CONFIGURABLE PATHS FOR DATABASES (see 'Databases' section of metaWRAP README for details)
# path to kraken standard database
# KRAKEN_DB=~/KRAKEN_DB
KRAKEN2_DB=~/database/kraken2.plus.plantandfungi
# path to indexed human (or other host) genome (see metaWRAP website for guide). This includes .bitmask and .srprism files
BMTAGGER_DB=~/database/human.genome
#
# paths to BLAST databases
BLASTDB=~/database/ncbi.nt
TAXDUMP=~/database/ncbi.taxonomy主要运行步骤
有一个自定义的脚本:这个脚本的作用是修改bins的编号,有时候编号太长了后面的步骤会报错。
import sys
fasta_file = sys.argv[1]
out_file = sys.argv[2]
index_file = sys.argv[3]
i = 0 # 从数字0开始
with open(fasta_file, "r") as f, open(out_file, "w") as out, open(index_file, "w") as index:
for line in f:
if line.startswith(">"):
i += 1
# 修改序列名称
out.write(">seq{0}\n".format(i))
# 输出新旧ID对应关系
old_id = line.replace(">","")
new_id = ">seq" + str(i)
index.write(str(new_id) + "\t" + old_id)
else:
out.write(line)
print("Done.")每一步程序:
# step.1:数据质控
for F in 0.data/*_1.fastq; do R=${F%_*}_2.fastq; BASE=${F##*/}; SAMPLE=${BASE%_*}; metawrap read_qc --skip-bmtagger -1 $F -2 $R -t 70 -o 1.read.qc/$SAMPLE & done
mkdir -p 2.clean.reads/all
for i in 1.read.qc/*; do
b=${i#*/}
mv ${i}/final_pure_reads_1.fastq 2.clean.reads/${b}_1.fastq
mv ${i}/final_pure_reads_2.fastq 2.clean.reads/${b}_2.fastq
done
# # step.2:合并所有序列用于组装
cat 2.clean.reads/*_1.fastq > 2.clean.reads/all/all_reads_1.fastq
cat 2.clean.reads/*_2.fastq > 2.clean.reads/all/all_reads_2.fastq
# step.3:开始组装
metawrap assembly -1 2.clean.reads/all/all_reads_1.fastq -2 2.clean.reads/all/all_reads_2.fastq -m 100 -t 60 --megahit -o 3.assembly
# step.4:Kraken2进行分类
metawrap kraken2 -o 4.kraken2 -s 1000000 2.clean.reads/*.fastq 3.assembly/final_assembly.fasta &
# step.5:开始分箱
metawrap binning -o 5.init.binning -t 60 -a 3.assembly/final_assembly.fasta --metabat2 --maxbin2 --concoct 2.clean.reads/*.fastq
# step.6:优化三个软件输出的分箱结果
metawrap bin_refinement -o 6.bin.refinement -t 60 -A 5.init.binning/metabat2_bins -B 5.init.binning/maxbin2_bins -C 5.init.binning/concoct_bins -c 50 -x 10
# step.7:分箱可视化
metawrap blobology -a 3.assembly/final_assembly.fasta -t 60 -o 7.blobology --bins 6.bin.refinement/metawrap_50_10_bins 2.clean.reads/*fastq
# step.8:分箱定量
metawrap quant_bins -b 6.bin.refinement/metawrap_50_10_bins -o 8.quant.bins -a 3.assembly/final_assembly.fasta 2.clean.reads/*fastq
# step.9:再次重新分箱,优化结果
metawrap reassemble_bins -o 9.bin.reassembly -1 2.clean.reads/all/all_reads_1.fastq -2 2.clean.reads/all/all_reads_2.fastq -t 60 -m 100 -c 50 -x 10 -b 6.bin.refinement/metawrap_50_10_bins
# step.10:分箱结果注释
metawrap classify_bins -b 9.bin.reassembly/reassembled_bins -o 10.bins.classification -t 60
# step.11:重命名bins的编号
mkdir 9.bin.reassembly/renamed.bins
mkdir 9.bin.reassembly/renamed.bins.id
for i in 9.bin.reassembly/reassembled_bins/*; do python3 ~/scripts/rename.fasta.id.py ${i} 9.bin.reassembly/renamed.bins/${i##*/} 9.bin.reassembly/renamed.bins.id/${i##*/}.id.txt; done
# step.12:注释bins
metawrap annotate_bins -o 11.bins.anno -t 60 -b 9.bin.reassembly/renamed.bins
# step.13:对bins的蛋白进行功能注释
# 这一步需要切换到python3环境下,直接调用python3的绝对路径也可以
mkdir 12.eggNOG-mapper
for i in 11.bins.anno/bin_translated_genes/*; do python3 ~/software/eggNOgmapper/emapper.py -m diamond --cpu 60 -i 11.bins.anno/bin_translated_genes/${i##*/} --output 12.eggNOG-mapper/${i##*/}.eggnogmapper.txt; doneMetaWRAP得到的是流程性的结果,个性化分析需要自行完成。
关于组装方法
宏基因组分析中最让人头疼的一个点就是组装的时候需要将所有样品的序列合并后再进行组装(因为某些基因的丰度非常低,单个样品组装的时候无法扫描到),这就需要大量的内存。现在宏基因组的数据量基本上是12G,左右两端加起来就有25G左右,再4个生物学重复,那就是100G了。随着样品数量增多,计算所需的内存数量也随之增加。这也就导致现在的宏基因组文章基本上是单样本组装的,也就是一个生物学重复进行一次组装。这个文章提出一种新的、对大多数研究者有用的组装方式:
改组装方法可以简单描述如下:
- 单个样本单独组装;
- 使用
BBnorm对原始数据进行标准化,相当于是随机抽取序列中的一部分,然后把所有数据合并后进行组装; - 将上述两种方法得到的组装结果合并,然后使用
MMSeqs对结果进行过滤。
NatCom文章方法
这个文章的分析思路比较有意思:
Sciecne文章方法
这篇文章真的是醍醐灌顶啊:
MAG dereplication
在这个文章中提到MAG dereplication,非常有用的:
Using the 459 MAGs that passed the minimum quality requirements, we dereplicated the MAGs using the “dereplicate” command of dRep (103) v3.3.0, performing the primary clustering at 90%, the secondary clustering at 95%, and requiring a coverage threshold (“nc”) of 30%.
MAG taxonomic
这个文章用了两种方法:
GTDBTKSGB:类似与系统发育树的方法检查MAG是否是之前报道过的。
群体遗传
在Jurdzinski K T, Mehrshad M, Delgado L F, et al. Large-scale phylogenomics of aquatic bacteria reveal molecular mechanisms for adaptation to salinity[J]. Science Advances, 2023, 9(21): eadg2059.看到POGENOM这个方法,找到了原文献:
DNA甲基化
机器学习方法
提取序列
ccsmeth call_hifi --subreads 01.data/bam/1.merged.bam --threads 60 --output 07.ccsmeth/01.callhifi/1.callhifi.bam序列比对
这一步的本质是用pbmm2进行比对 。
ccsmeth align_hifi --hifireads 01.data/bam/1.merged.bam --ref 01.data/ref/r498.fa --output 07.ccsmeth/02.alignhifi/1.align.hifi.pbmm2.bam --threads 60变异检测
CUDA_VISIBLE_DEVICES=0 ccsmeth call_mods --input 03.mapping/02.bam/1.bam --re 01.data/ref/r498.fa --model_file 01.data/model_ccsmeth_5mCpG_call_mods_attbigru2s_b21.v2.ckpt --output 07.ccsmeth/03.callmodification/1.callmods --threads 60 --threads_call 60 --model_type attbigru2s --mode align变异频率
````
### BiaMark流程
#### 基因组准备
````sh
bismark_genome_preparation --path_to_aligner ~/mambaforge/envs/BisMark/bin/ --verbose 06.CpG.methylation.PC/03.bismark.index/ BisMark
bismark -q -p 60 --score_min L,0,-0.6 -X 1000 ./06.CpG.methylation.PC/03.bismark.index -1 ./06.CpG.methylation.PC/01.clean.data/CRR190475_f1.fq.gz -2 ./06.CpG.methylation.PC/01.clean.data/CRR190475_r2.fq.gz -o 06.CpG.methylation.PC/04.bismark.bam


